 Suche
Suche
 Mein Konto
Mein Konto
Retinoblastoma
Retinoblastoma
descripción general
Anatomía del ojo
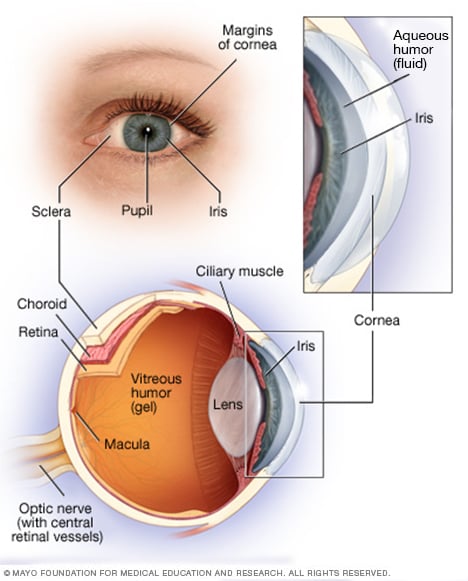
Anatomía del ojo
Su ojo es una estructura compleja y compacta de unos 2,5 cm de diámetro. Recibe millones de datos sobre el mundo exterior, que el cerebro procesa rápidamente.
El retinoblastoma es un cáncer de ojo que comienza en la retina, el delicado revestimiento del interior del ojo. El retinoblastoma afecta con mayor frecuencia a niños pequeños, pero rara vez ocurre en adultos.
La retina está formada por tejido nervioso que detecta la luz cuando pasa por la parte frontal del ojo. La retina envía señales a través del nervio óptico al cerebro, donde estas señales se interpretan como imágenes.
El retinoblastoma es una forma rara de cáncer de ojo y la forma más común de cáncer que afecta el ojo en los niños. El retinoblastoma puede ocurrir en uno o ambos ojos.
Síntomas
Dado que el retinoblastoma afecta principalmente a bebés y niños pequeños, los síntomas no son comunes. Los signos que puede notar incluyen:
- Eine weiße Farbe im mittleren Augenkreis (Pupille), wenn Licht in das Auge scheint, z. B. wenn jemand ein Blitzfoto des Kindes macht
- Augen, die in verschiedene Richtungen zu schauen scheinen
- Schlechte Sicht
- Augenrötung
- Augenschwellung
¿Cuándo acudir al médico?
Concierte una cita con el médico de su hijo si nota algún cambio en los ojos de su hijo que le preocupe. El retinoblastoma es un cáncer poco común, por lo que el médico de su hijo puede examinar primero otras enfermedades oculares más comunes.
Si tiene antecedentes familiares de retinoblastoma, hable con su médico si planea tener hijos.
Causas
Patrón de herencia autosómico dominante
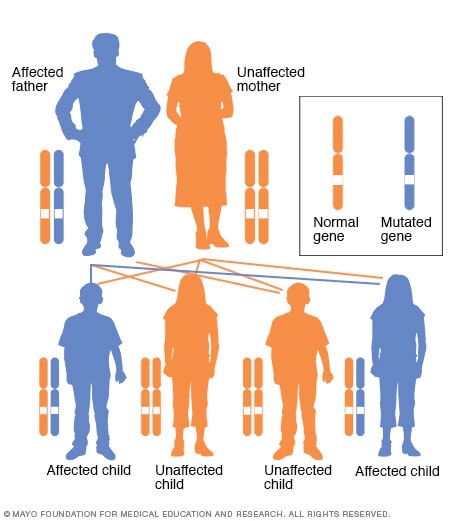
Patrón de herencia autosómico dominante
En un trastorno autosómico dominante, el gen alterado es un gen dominante ubicado en uno de los cromosomas no sexuales (autosomas). Sólo se necesita un gen alterado para verse afectado por este tipo de trastorno. Una persona con un trastorno autosómico dominante (en este caso el padre) tiene un 50 por ciento de posibilidades de tener un hijo afectado con un gen alterado (gen dominante) y un 50 por ciento de posibilidades de tener un hijo no afectado con dos genes típicos (genes recesivos).
El retinoblastoma ocurre cuando las células nerviosas de la retina desarrollan mutaciones genéticas. Estas mutaciones hacen que las células sigan creciendo y multiplicándose cuando las células sanas morirían. Esta masa celular acumulada forma un tumor.
Las células de retinoblastoma pueden invadir más el ojo y las estructuras cercanas. El retinoblastoma también puede diseminarse (hacer metástasis) a otras áreas del cuerpo, incluidos el cerebro y la columna.
En la mayoría de los casos de retinoblastoma, no está claro qué causa la mutación genética que provoca el cáncer. Sin embargo, es posible que los niños hereden una mutación genética de sus padres.
Retinoblastoma, que se hereda
Las mutaciones genéticas que aumentan el riesgo de retinoblastoma y otros cánceres pueden transmitirse de padres a hijos.
El retinoblastoma hereditario se transmite de padres a hijos de forma autosómica dominante, lo que significa que sólo uno de los padres necesita una única copia del gen mutado para transmitir el mayor riesgo de retinoblastoma a los niños. Si uno de los padres es portador de un gen mutado, cada hijo tiene un 50 por ciento de posibilidades de heredar ese gen.
Aunque una mutación genética aumenta el riesgo de que un niño sufra retinoblastoma, no significa que el cáncer sea inevitable.
Los niños con la forma hereditaria de retinoblastoma tienden a desarrollar la enfermedad a una edad más temprana. El retinoblastoma hereditario también tiende a aparecer en ambos ojos, en lugar de en un solo ojo.
Complicaciones
Los niños tratados por retinoblastoma corren el riesgo de que el cáncer regrese dentro y alrededor del ojo tratado. Por este motivo, el médico de su hijo programará visitas de seguimiento para detectar retinoblastoma recurrente. El médico puede crear un programa de atención de seguimiento personalizado para su hijo que incluya exámenes oculares frecuentes.
Además, los niños con la forma hereditaria de retinoblastoma tienen un mayor riesgo de desarrollar otros cánceres en cualquier parte del cuerpo en los años posteriores al tratamiento, especialmente pineoblastoma, un tipo de tumor cerebral. Por esta razón, los niños con retinoblastoma hereditario pueden ser examinados periódicamente para detectar otros cánceres.
prevención
Los médicos no están seguros de qué causa la mayoría de los casos de retinoblastoma, por lo que no existe una forma comprobada de prevenir la enfermedad.
Prevención para familias con retinoblastoma hereditario
Si a su hijo le diagnostican retinoblastoma, su médico puede recomendarle pruebas genéticas para determinar si el cáncer fue causado por una mutación genética hereditaria. Su médico puede recomendarle que se reúna con un asesor genético que pueda ayudarlo a decidir si debe someterse a pruebas genéticas.
Las pruebas genéticas permiten a las familias saber si sus hijos pueden tener un mayor riesgo de padecer retinoblastoma para poder planificar la atención médica en consecuencia. Por ejemplo, los exámenes de la vista pueden comenzar poco después del nacimiento o, en algunas situaciones, antes de que nazca el bebé. De esta manera, el retinoblastoma se puede diagnosticar muy temprano, cuando el tumor es pequeño y todavía existe la posibilidad de curarse y preservar la visión.
Las pruebas genéticas se pueden utilizar para determinar si:
- Ihr Kind mit Retinoblastom hat ein Risiko für andere verwandte Krebsarten.
- Ihr Kind mit Retinoblastom kann eine Genmutation tragen, die an seine zukünftigen Kinder weitergegeben werden kann.
- Ihre anderen Kinder sind einem Retinoblastom und anderen verwandten Krebsarten ausgesetzt.
- Sie und Ihr Partner haben die Möglichkeit, die genetische Mutation an zukünftige Kinder weiterzugeben.
Fuentes:
- Retinoblastom-Behandlung (PDQ) – Version für medizinische Fachkräfte. Nationales Krebs Institut. https://www.cancer.gov/types/retinoblastoma/hp/retinoblastoma-treatment-pdq. Abgerufen am 18. Januar 2021.
- Yanoff M. et al., Hrsg. Bösartige intraokulare Neoplasien. In: Augenheilkunde. 5. Aufl.: Elsevier; 2019. https://www.clinicalkey.com. Abgerufen am 18. Januar 2021.
- Chirurgische Maßnahmen. American Society of Ocularists. https://www.ocularist.org/resources_surgical_procedures.asp. Abgerufen am 21. Januar 2021.
- Kliegman RM, et al., Hrsg. Retinoblastom In: Nelson Textbook of Pediatrics. 21. Aufl. Elsevier; 2020. https://www.clinicalkey.com. Abgerufen am 4. Dezember 2020.
- Dimaras H. et al. Retinoblastom, der sichtbare ZNS-Tumor: Eine Übersicht. Zeitschrift für neurowissenschaftliche Forschung. 2019; doi:10.1002/jnr.24213.
- Fragen Sie MayoExpert. Retinoblastom (Kind). Mayo-Klinik; 2019.
- Hartnett ME, Hrsg. Pädiatrische Netzhaut. 3. Aufl. Lippincott Williams & Wilkins; 2020.