 Suche
Suche
 Mein Konto
Mein Konto
Retinoblastoma
Retinoblastoma
apžvalga
Akies anatomija
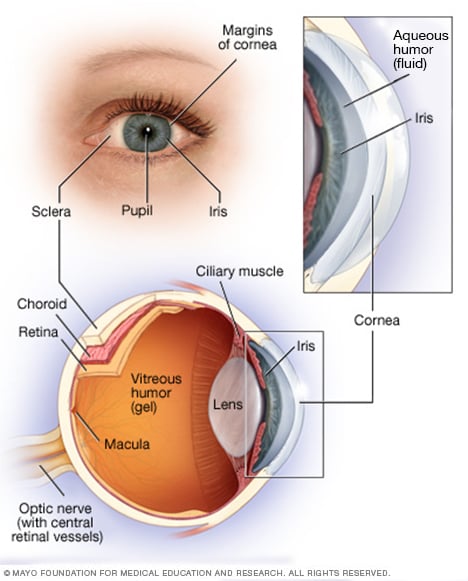
Akies anatomija
Jūsų akis yra sudėtinga ir kompaktiška struktūra, kurios skersmuo yra apie 2,5 cm. Jis gauna milijonus informacijos apie išorinį pasaulį, kurią greitai apdoroja jūsų smegenys.
Retinoblastoma yra akių vėžys, kuris prasideda tinklainėje – subtilioje akies gleivinėje. Retinoblastoma dažniausiai paveikia mažus vaikus, tačiau retai gali pasireikšti suaugusiems.
Jūsų tinklainė sudaryta iš nervinio audinio, kuris jaučia šviesą, kai ji patenka per priekinę akies dalį. Tinklainė siunčia signalus per jūsų regos nervą į jūsų smegenis, kur šie signalai interpretuojami kaip vaizdai.
Retinoblastoma yra reta akių vėžio forma ir dažniausia vaikų akis pažeidžianti vėžio forma. Retinoblastoma gali atsirasti vienoje arba abiejose akyse.
Simptomai
Kadangi retinoblastoma pirmiausia paveikia kūdikius ir mažus vaikus, simptomai nėra dažni. Ženklai, kuriuos galite pastebėti, yra šie:
- Eine weiße Farbe im mittleren Augenkreis (Pupille), wenn Licht in das Auge scheint, z. B. wenn jemand ein Blitzfoto des Kindes macht
- Augen, die in verschiedene Richtungen zu schauen scheinen
- Schlechte Sicht
- Augenrötung
- Augenschwellung
Kada eiti pas gydytoja?
Pasitarkite su savo vaiko gydytoju, jei pastebėsite kokių nors jums rūpimų vaiko akių pokyčių. Retinoblastoma yra retas vėžys, todėl jūsų vaiko gydytojas pirmiausia gali patikrinti kitas dažniau pasitaikančias akių ligas.
Jei jūsų šeimoje yra buvę retinoblastomos atvejų, pasitarkite su gydytoju, jei planuojate turėti vaikų.
Priežastys
Autosominis dominuojantis paveldėjimo modelis
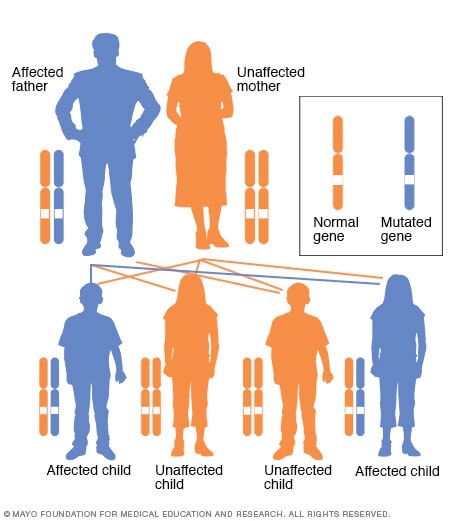
Autosominis dominuojantis paveldėjimo modelis
Sergant autosominiu dominuojančiu sutrikimu, pakitęs genas yra dominuojantis genas, esantis vienoje iš nelytinių chromosomų (autosomų). Jums reikia tik vieno pakeisto geno, kad galėtumėte paveikti tokio tipo sutrikimą. Asmuo, turintis autosominį dominuojantį sutrikimą – šiuo atveju tėvas – turi 50 procentų tikimybę, kad susilauks paveikto vaiko su vienu pakitusiu genu (dominuojančiu genu), ir 50 procentų tikimybę, kad vaikas turės nepakitusio vaiko su dviem tipiniais genais (recesyviniais genais).
Retinoblastoma atsiranda, kai tinklainės nervinėse ląstelėse išsivysto genetinės mutacijos. Dėl šių mutacijų ląstelės toliau auga ir dauginasi, kai sveikos ląstelės miršta. Ši besikaupianti ląstelių masė formuoja naviką.
Retinoblastomos ląstelės gali prasiskverbti toliau į akį ir netoliese esančias struktūras. Retinoblastoma taip pat gali plisti (metastazuoti) į kitas kūno vietas, įskaitant smegenis ir stuburą.
Daugeliu retinoblastomos atvejų neaišku, kas sukelia genetinę mutaciją, sukeliančią vėžį. Tačiau vaikai gali paveldėti genetinę mutaciją iš savo tėvų.
Retinoblastoma, kuri yra paveldima
Genų mutacijos, didinančios retinoblastomos ir kitų vėžio formų riziką, gali būti perduodamos iš tėvų vaikams.
Paveldima retinoblastoma iš tėvų perduodama vaikams autosominiu dominuojančiu būdu, o tai reiškia, kad tik vienam iš tėvų reikia vienos mutavusio geno kopijos, kad padidintų retinoblastomos riziką vaikams. Jei vienas iš tėvų turi mutavusį geną, kiekvienas vaikas turi 50 procentų galimybę paveldėti tą geną.
Nors genetinė mutacija padidina vaiko retinoblastomos riziką, tai nereiškia, kad vėžys yra neišvengiamas.
Vaikai, sergantys paveldima retinoblastomos forma, dažniausiai suserga anksčiau. Paveldima retinoblastoma taip pat linkusi atsirasti abiem akimis, o ne tik viena.
Komplikacijos
Vaikams, gydytiems nuo retinoblastomos, yra rizika, kad vėžys atsinaujins gydomoje akyje ir aplink ją. Dėl šios priežasties jūsų vaiko gydytojas paskirs tolesnius vizitus, kad patikrintų, ar nėra pasikartojančios retinoblastomos. Gydytojas gali sudaryti asmeninį jūsų vaiko priežiūros tvarkaraštį, kuris apima dažnus akių tyrimus.
Be to, vaikai, sergantys paveldima retinoblastomos forma, turi didesnę riziką susirgti kitomis vėžio formomis bet kurioje kūno vietoje per kelerius metus po gydymo, ypač pineoblastomos, tam tikros rūšies smegenų auglio. Dėl šios priežasties vaikai, sergantys paveldima retinoblastoma, gali būti reguliariai tikrinami, siekiant patikrinti, ar nėra kitų vėžio formų.
prevencija
Gydytojai nėra tikri, kas sukelia daugumą retinoblastomos atvejų, todėl nėra įrodyto būdo užkirsti kelią ligai.
Prevencija šeimoms, turinčioms paveldimą retinoblastomą
Jei jūsų vaikui diagnozuota retinoblastoma, gydytojas gali rekomenduoti atlikti genetinį tyrimą, kad nustatytų, ar vėžį sukėlė paveldėta genų mutacija. Jūsų gydytojas gali rekomenduoti susitikti su genetikos konsultantu, kuris gali padėti nuspręsti, ar jums reikia atlikti genetinį tyrimą.
Genetiniai tyrimai leidžia šeimoms sužinoti, ar jų vaikams gali būti padidėjusi retinoblastomos rizika, kad būtų galima atitinkamai planuoti medicininę priežiūrą. Pavyzdžiui, akių tyrimai gali prasidėti netrukus po gimimo arba, kai kuriais atvejais, prieš kūdikio gimimą. Tokiu būdu retinoblastomą galima diagnozuoti labai anksti – kai auglys nedidelis ir dar yra galimybė pasveikti bei išsaugoti regėjimą.
Genetiniai tyrimai gali būti naudojami siekiant nustatyti, ar:
- Ihr Kind mit Retinoblastom hat ein Risiko für andere verwandte Krebsarten.
- Ihr Kind mit Retinoblastom kann eine Genmutation tragen, die an seine zukünftigen Kinder weitergegeben werden kann.
- Ihre anderen Kinder sind einem Retinoblastom und anderen verwandten Krebsarten ausgesetzt.
- Sie und Ihr Partner haben die Möglichkeit, die genetische Mutation an zukünftige Kinder weiterzugeben.
Šaltiniai:
- Retinoblastom-Behandlung (PDQ) – Version für medizinische Fachkräfte. Nationales Krebs Institut. https://www.cancer.gov/types/retinoblastoma/hp/retinoblastoma-treatment-pdq. Abgerufen am 18. Januar 2021.
- Yanoff M. et al., Hrsg. Bösartige intraokulare Neoplasien. In: Augenheilkunde. 5. Aufl.: Elsevier; 2019. https://www.clinicalkey.com. Abgerufen am 18. Januar 2021.
- Chirurgische Maßnahmen. American Society of Ocularists. https://www.ocularist.org/resources_surgical_procedures.asp. Abgerufen am 21. Januar 2021.
- Kliegman RM, et al., Hrsg. Retinoblastom In: Nelson Textbook of Pediatrics. 21. Aufl. Elsevier; 2020. https://www.clinicalkey.com. Abgerufen am 4. Dezember 2020.
- Dimaras H. et al. Retinoblastom, der sichtbare ZNS-Tumor: Eine Übersicht. Zeitschrift für neurowissenschaftliche Forschung. 2019; doi:10.1002/jnr.24213.
- Fragen Sie MayoExpert. Retinoblastom (Kind). Mayo-Klinik; 2019.
- Hartnett ME, Hrsg. Pädiatrische Netzhaut. 3. Aufl. Lippincott Williams & Wilkins; 2020.