 Suche
Suche
 Mein Konto
Mein Konto
Retinoblastoom
Retinoblastoom
overzicht
Anatomie van het oog
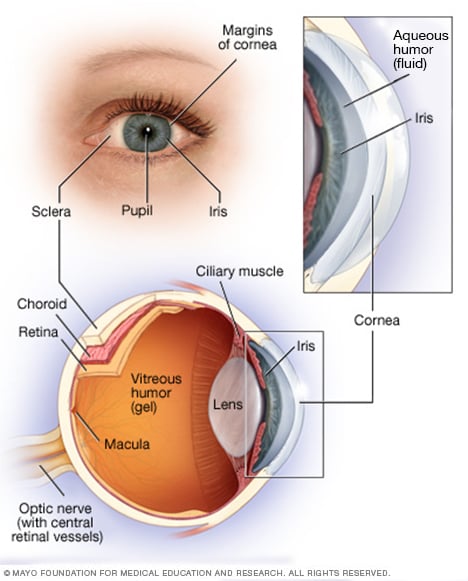
Anatomie van het oog
Je oog is een complexe en compacte structuur met een diameter van ongeveer 2,5 cm. Het ontvangt miljoenen stukjes informatie over de buitenwereld, die snel door je hersenen worden verwerkt.
Retinoblastoom is een oogkanker die begint in het netvlies – het delicate slijmvlies in uw oog. Retinoblastoom treft meestal jonge kinderen, maar kan zelden voorkomen bij volwassenen.
Uw netvlies bestaat uit zenuwweefsel dat licht waarneemt als het door de voorkant van uw oog komt. Het netvlies stuurt signalen via uw oogzenuw naar uw hersenen, waar deze signalen worden geïnterpreteerd als beelden.
Retinoblastoom is een zeldzame vorm van oogkanker en de meest voorkomende vorm van kanker aan het oog bij kinderen. Retinoblastoom kan in één of beide ogen voorkomen.
Symptomen
Omdat retinoblastoom vooral zuigelingen en jonge kinderen treft, komen de symptomen niet vaak voor. Tekenen die u mogelijk opmerkt zijn onder meer:
- Eine weiße Farbe im mittleren Augenkreis (Pupille), wenn Licht in das Auge scheint, z. B. wenn jemand ein Blitzfoto des Kindes macht
- Augen, die in verschiedene Richtungen zu schauen scheinen
- Schlechte Sicht
- Augenrötung
- Augenschwellung
Wanneer naar de dokter?
Maak een afspraak met de arts van uw kind als u veranderingen in de ogen van uw kind opmerkt die u zorgen baren. Retinoblastoom is een zeldzame vorm van kanker, dus de arts van uw kind kan eerst andere, vaker voorkomende oogziekten controleren.
Als u een familiegeschiedenis van retinoblastoom heeft, bespreek dit dan met uw arts als u van plan bent kinderen te krijgen.
Oorzaken
Autosomaal dominant overervingspatroon
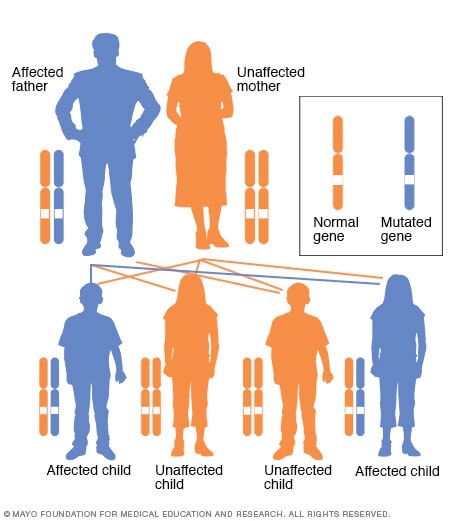
Autosomaal dominant overervingspatroon
Bij een autosomaal dominante aandoening is het veranderde gen een dominant gen dat zich op een van de niet-geslachtschromosomen (autosomen) bevindt. Er is maar één veranderd gen nodig om door dit type aandoening getroffen te worden. Een persoon met een autosomaal dominante aandoening – in dit geval de vader – heeft 50 procent kans op een getroffen kind met één veranderd gen (dominant gen) en 50 procent kans op een onaangetast kind met twee typische genen (recessieve genen).
Retinoblastoom treedt op wanneer zenuwcellen in het netvlies genetische mutaties ontwikkelen. Deze mutaties zorgen ervoor dat cellen blijven groeien en zich vermenigvuldigen wanneer gezonde cellen zouden afsterven. Deze ophopende celmassa vormt een tumor.
Retinoblastoomcellen kunnen verder in het oog en nabijgelegen structuren binnendringen. Retinoblastoom kan zich ook verspreiden (uitzaaien) naar andere delen van het lichaam, inclusief de hersenen en de wervelkolom.
In de meeste gevallen van retinoblastoom is het niet duidelijk wat de genetische mutatie veroorzaakt die tot kanker leidt. Het is echter mogelijk dat kinderen een genetische mutatie van hun ouders erven.
Retinoblastoom, dat erfelijk is
Genmutaties die het risico op retinoblastoom en andere vormen van kanker verhogen, kunnen van ouders op kinderen worden overgedragen.
Erfelijk retinoblastoom wordt op autosomaal dominante wijze doorgegeven van ouders op kinderen, wat betekent dat slechts één ouder één kopie van het gemuteerde gen nodig heeft om het verhoogde risico op retinoblastoom door te geven aan de kinderen. Als een ouder een gemuteerd gen draagt, heeft elk kind een kans van 50 procent om dat gen te erven.
Hoewel een genetische mutatie het risico van een kind op retinoblastoom vergroot, betekent dit niet dat kanker onvermijdelijk is.
Kinderen met de erfelijke vorm van retinoblastoom ontwikkelen de ziekte doorgaans op jongere leeftijd. Erfelijk retinoblastoom komt ook vaak in beide ogen voor, in tegenstelling tot slechts één oog.
Complicaties
Kinderen die voor retinoblastoom worden behandeld, lopen het risico dat de kanker terugkeert in en rond het behandelde oog. Om deze reden zal de arts van uw kind vervolgbezoeken plannen om te controleren op recidiverend retinoblastoom. De arts kan een persoonlijk nazorgschema voor uw kind opstellen, inclusief frequente oogonderzoeken.
Bovendien hebben kinderen met de erfelijke vorm van retinoblastoom een verhoogd risico op het ontwikkelen van andere vormen van kanker in welk deel van het lichaam dan ook in de jaren na de behandeling, vooral pineoblastoom, een type hersentumor. Om deze reden kunnen kinderen met erfelijk retinoblastoom regelmatig worden gescreend om te controleren op andere vormen van kanker.
preventie
Artsen weten niet zeker wat de meeste gevallen van retinoblastoom veroorzaakt, dus er is geen bewezen manier om de ziekte te voorkomen.
Preventie voor gezinnen met erfelijk retinoblastoom
Als bij uw kind de diagnose retinoblastoom is gesteld, kan uw arts genetische tests aanbevelen om vast te stellen of de kanker werd veroorzaakt door een erfelijke genmutatie. Uw arts kan u aanraden een genetisch adviseur te raadplegen die u kan helpen beslissen of u een genetische test moet ondergaan.
Dankzij genetische tests kunnen gezinnen weten of hun kinderen mogelijk een verhoogd risico lopen op retinoblastoom, zodat de medische zorg dienovereenkomstig kan worden gepland. Oogonderzoeken kunnen bijvoorbeeld kort na de geboorte beginnen of, in sommige situaties, voordat een baby wordt geboren. Op deze manier kan retinoblastoom zeer vroeg worden gediagnosticeerd - wanneer de tumor klein is en er nog steeds een kans is op genezing en behoud van het gezichtsvermogen.
Genetische tests kunnen worden gebruikt om te bepalen of:
- Ihr Kind mit Retinoblastom hat ein Risiko für andere verwandte Krebsarten.
- Ihr Kind mit Retinoblastom kann eine Genmutation tragen, die an seine zukünftigen Kinder weitergegeben werden kann.
- Ihre anderen Kinder sind einem Retinoblastom und anderen verwandten Krebsarten ausgesetzt.
- Sie und Ihr Partner haben die Möglichkeit, die genetische Mutation an zukünftige Kinder weiterzugeben.
Bronnen:
- Retinoblastom-Behandlung (PDQ) – Version für medizinische Fachkräfte. Nationales Krebs Institut. https://www.cancer.gov/types/retinoblastoma/hp/retinoblastoma-treatment-pdq. Abgerufen am 18. Januar 2021.
- Yanoff M. et al., Hrsg. Bösartige intraokulare Neoplasien. In: Augenheilkunde. 5. Aufl.: Elsevier; 2019. https://www.clinicalkey.com. Abgerufen am 18. Januar 2021.
- Chirurgische Maßnahmen. American Society of Ocularists. https://www.ocularist.org/resources_surgical_procedures.asp. Abgerufen am 21. Januar 2021.
- Kliegman RM, et al., Hrsg. Retinoblastom In: Nelson Textbook of Pediatrics. 21. Aufl. Elsevier; 2020. https://www.clinicalkey.com. Abgerufen am 4. Dezember 2020.
- Dimaras H. et al. Retinoblastom, der sichtbare ZNS-Tumor: Eine Übersicht. Zeitschrift für neurowissenschaftliche Forschung. 2019; doi:10.1002/jnr.24213.
- Fragen Sie MayoExpert. Retinoblastom (Kind). Mayo-Klinik; 2019.
- Hartnett ME, Hrsg. Pädiatrische Netzhaut. 3. Aufl. Lippincott Williams & Wilkins; 2020.