 Suche
Suche
 Mein Konto
Mein Konto
Retinoblastom
Retinoblastom
oversikt
Anatomi av øyet
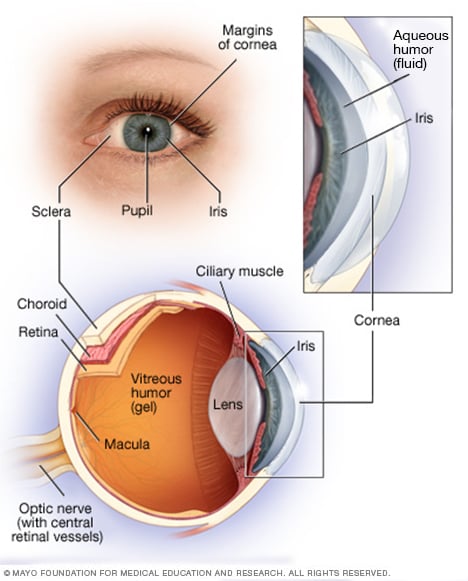
Anatomi av øyet
Øyet ditt er en kompleks og kompakt struktur med en diameter på omtrent 2,5 cm. Den mottar millioner av informasjon om omverdenen, som raskt behandles av hjernen din.
Retinoblastom er en øyekreft som begynner i netthinnen - den delikate slimhinnen inne i øyet. Retinoblastom rammer oftest små barn, men kan sjelden forekomme hos voksne.
Netthinnen din består av nervevev som registrerer lys når det kommer gjennom fronten av øyet. Netthinnen sender signaler gjennom synsnerven til hjernen din, hvor disse signalene tolkes som bilder.
Retinoblastom er en sjelden form for øyekreft og den vanligste kreftformen som rammer øyet hos barn. Retinoblastom kan forekomme i ett eller begge øyne.
Symptomer
Fordi retinoblastom først og fremst rammer spedbarn og små barn, er symptomene ikke vanlige. Tegn du kan legge merke til inkluderer:
- Eine weiße Farbe im mittleren Augenkreis (Pupille), wenn Licht in das Auge scheint, z. B. wenn jemand ein Blitzfoto des Kindes macht
- Augen, die in verschiedene Richtungen zu schauen scheinen
- Schlechte Sicht
- Augenrötung
- Augenschwellung
Når skal man gå til legen?
Bestill en avtale med barnets lege hvis du merker endringer i barnets øyne som angår deg. Retinoblastom er en sjelden kreft, så barnets lege kan sjekke andre mer vanlige øyesykdommer først.
Hvis du har en familiehistorie med retinoblastom, diskuter dette med legen din hvis du planlegger å få barn.
Årsaker
Autosomalt dominant arvemønster
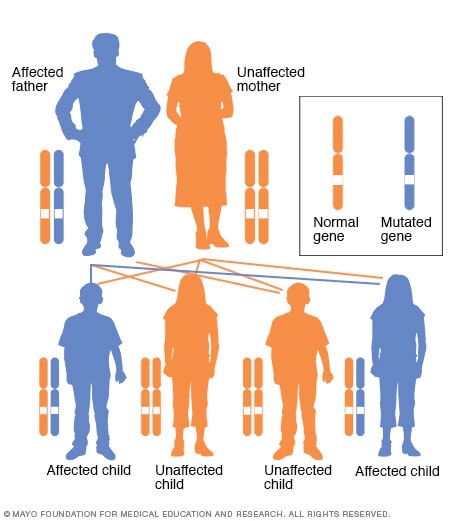
Autosomalt dominant arvemønster
I en autosomal dominant lidelse er det endrede genet et dominant gen som ligger på et av de ikke-kjønnslige kromosomene (autosomene). Du trenger bare ett endret gen for å bli påvirket av denne typen lidelse. En person med en autosomal dominant lidelse – i dette tilfellet faren – har 50 prosent sjanse for å få et affisert barn med ett endret gen (dominant gen) og 50 prosent sjanse for å få et upåvirket barn med to typiske gener (recessive gener).
Retinoblastom oppstår når nerveceller i netthinnen utvikler genetiske mutasjoner. Disse mutasjonene får celler til å fortsette å vokse og formere seg når friske celler dør. Denne akkumulerende cellemassen danner en svulst.
Retinoblastomceller kan invadere lenger inn i øyet og nærliggende strukturer. Retinoblastom kan også spre seg (metastasere) til andre områder av kroppen, inkludert hjernen og ryggraden.
I de fleste tilfeller av retinoblastom er det ikke klart hva som forårsaker den genetiske mutasjonen som fører til kreft. Det er imidlertid mulig for barn å arve en genetisk mutasjon fra foreldrene.
Retinoblastoma, som er arvelig
Genmutasjoner som øker risikoen for retinoblastom og andre kreftformer kan overføres fra foreldre til barn.
Arvelig retinoblastom overføres fra foreldre til barn på en autosomalt dominant måte, noe som betyr at bare én forelder trenger en enkelt kopi av det muterte genet for å overføre den økte risikoen for retinoblastom til barna. Hvis en forelder bærer et mutert gen, har hvert barn 50 prosent sjanse for å arve det genet.
Selv om en genetisk mutasjon øker et barns risiko for retinoblastom, betyr det ikke at kreft er uunngåelig.
Barn med den arvelige formen for retinoblastom har en tendens til å utvikle sykdommen i en tidligere alder. Arvelig retinoblastom har også en tendens til å forekomme i begge øyne, i motsetning til bare ett øye.
Komplikasjoner
Barn som behandles for retinoblastom risikerer at kreften kommer tilbake i og rundt det behandlede øyet. Av denne grunn vil barnets lege planlegge oppfølgingsbesøk for å se etter tilbakevendende retinoblastom. Legen kan lage en personlig oppfølgingsplan for barnet ditt som inkluderer hyppige øyeundersøkelser.
I tillegg har barn med den arvelige formen for retinoblastom en økt risiko for å utvikle andre kreftformer i hvilken som helst del av kroppen i årene etter behandlingen, spesielt pineoblastom, en type hjernesvulst. Av denne grunn kan barn med arvelig retinoblastom screenes regelmessig for å se etter andre kreftformer.
forebygging
Leger er ikke sikre på hva som forårsaker de fleste tilfeller av retinoblastom, så det er ingen påvist måte å forhindre sykdommen på.
Forebygging for familier med arvelig retinoblastom
Hvis barnet ditt er diagnostisert med retinoblastom, kan legen din anbefale genetisk testing for å avgjøre om kreften var forårsaket av en arvelig genmutasjon. Legen din kan anbefale at du møter en genetisk rådgiver som kan hjelpe deg med å bestemme om du bør gjennomgå genetisk testing.
Genetisk testing lar familier vite om barna deres kan ha økt risiko for retinoblastom, slik at medisinsk behandling kan planlegges deretter. For eksempel kan øyeundersøkelser begynne kort tid etter fødselen eller, i noen situasjoner, før en baby er født. På denne måten kan retinoblastom diagnostiseres veldig tidlig – når svulsten er liten og det fortsatt er mulighet for å helbrede og bevare synet.
Genetisk testing kan brukes til å avgjøre om:
- Ihr Kind mit Retinoblastom hat ein Risiko für andere verwandte Krebsarten.
- Ihr Kind mit Retinoblastom kann eine Genmutation tragen, die an seine zukünftigen Kinder weitergegeben werden kann.
- Ihre anderen Kinder sind einem Retinoblastom und anderen verwandten Krebsarten ausgesetzt.
- Sie und Ihr Partner haben die Möglichkeit, die genetische Mutation an zukünftige Kinder weiterzugeben.
Kilder:
- Retinoblastom-Behandlung (PDQ) – Version für medizinische Fachkräfte. Nationales Krebs Institut. https://www.cancer.gov/types/retinoblastoma/hp/retinoblastoma-treatment-pdq. Abgerufen am 18. Januar 2021.
- Yanoff M. et al., Hrsg. Bösartige intraokulare Neoplasien. In: Augenheilkunde. 5. Aufl.: Elsevier; 2019. https://www.clinicalkey.com. Abgerufen am 18. Januar 2021.
- Chirurgische Maßnahmen. American Society of Ocularists. https://www.ocularist.org/resources_surgical_procedures.asp. Abgerufen am 21. Januar 2021.
- Kliegman RM, et al., Hrsg. Retinoblastom In: Nelson Textbook of Pediatrics. 21. Aufl. Elsevier; 2020. https://www.clinicalkey.com. Abgerufen am 4. Dezember 2020.
- Dimaras H. et al. Retinoblastom, der sichtbare ZNS-Tumor: Eine Übersicht. Zeitschrift für neurowissenschaftliche Forschung. 2019; doi:10.1002/jnr.24213.
- Fragen Sie MayoExpert. Retinoblastom (Kind). Mayo-Klinik; 2019.
- Hartnett ME, Hrsg. Pädiatrische Netzhaut. 3. Aufl. Lippincott Williams & Wilkins; 2020.