DiGeorge-Syndrom (22q11.2-Deletionssyndrom)

Überblick
Das DiGeorge-Syndrom, besser bekannt unter einem breiteren Begriff – 22q11.2-Deletionssyndrom – ist eine Störung, die verursacht wird, wenn ein kleiner Teil des Chromosoms 22 fehlt. Diese Deletion führt zu einer schlechten Entwicklung mehrerer Körpersysteme.
Der Begriff 22q11.2-Deletionssyndrom umfasst Begriffe, die einst als separate Zustände angesehen wurden, einschließlich DiGeorge-Syndrom, Velokardiofaziales Syndrom und andere Erkrankungen, die dieselbe genetische Ursache haben, obwohl die Merkmale leicht variieren können.
Zu den medizinischen Problemen, die häufig mit dem 22q11.2-Deletionssyndrom in Verbindung gebracht werden, gehören Herzfehler, eine schlechte Funktion des Immunsystems, eine Gaumenspalte, Komplikationen im Zusammenhang mit einem niedrigen Kalziumspiegel im Blut und eine verzögerte Entwicklung mit Verhaltens- und emotionalen Problemen.
Die Anzahl und Schwere der mit dem 22q11.2-Deletionssyndrom verbundenen Symptome variieren. Fast jeder mit diesem Syndrom benötigt jedoch eine Behandlung durch Spezialisten auf verschiedenen Gebieten.
Symptome
Anzeichen und Symptome des DiGeorge-Syndroms (22q11.2-Deletionssyndrom) können in Art und Schweregrad variieren, je nachdem, welche Körpersysteme betroffen sind und wie schwer die Defekte sind. Einige Anzeichen und Symptome können bei der Geburt sichtbar sein, andere treten möglicherweise erst später im Säuglingsalter oder in der frühen Kindheit auf.
Anzeichen und Symptome können eine Kombination der folgenden sein:
- Herzgeräusch und bläuliche Haut aufgrund schlechter Zirkulation von sauerstoffreichem Blut (Zyanose) als Folge eines Herzfehlers
- Häufige Infektionen
- Bestimmte Gesichtszüge, wie ein unterentwickeltes Kinn, tief angesetzte Ohren, weit auseinanderstehende Augen oder eine schmale Furche in der Oberlippe
- Eine Lücke im Gaumen (Gaumenspalte) oder andere Probleme mit dem Gaumen
- Verzögertes Wachstum
- Schwierigkeiten bei der Nahrungsaufnahme, Gewichtszunahme oder Magen-Darm-Probleme
- Atembeschwerden
- Schlechter Muskeltonus
- Verzögerte Entwicklung, wie z. B. Verzögerungen beim Umdrehen, Aufsetzen oder anderen kindlichen Meilensteinen
- Verzögerte Sprachentwicklung oder nasal klingende Sprache
- Lernverzögerungen oder Behinderungen
- Verhaltensprobleme
Wann zum arzt
Andere Erkrankungen können ähnliche Anzeichen und Symptome wie das 22q11.2-Deletionssyndrom verursachen. Daher ist es wichtig, eine genaue und schnelle Diagnose zu erhalten, wenn Ihr Kind die oben aufgeführten Anzeichen oder Symptome zeigt.
Ärzte können ein 22q11.2-Deletionssyndrom vermuten:
- Bei der Geburt. Wenn bestimmte Zustände – ein schwerer Herzfehler, eine Gaumenspalte oder eine Kombination anderer Faktoren, die für das 22q11.2-Deletionssyndrom typisch sind – bereits bei der Geburt offensichtlich sind, werden diagnostische Tests wahrscheinlich beginnen, bevor Ihr Kind das Krankenhaus verlässt.
- Bei wohlauf-Baby-Besuchen. Ihr Hausarzt oder Kinderarzt kann die Störung aufgrund einer Kombination von Krankheiten oder Störungen vermuten, die im Laufe der Zeit auftreten. Andere Probleme können Ihrem Arzt während regelmäßig geplanter Arztbesuche oder jährlicher Vorsorgeuntersuchungen für Ihr Kind auffallen.
Ursachen
Jede Person hat zwei Kopien von Chromosom 22, eine von jedem Elternteil geerbt. Wenn eine Person das DiGeorge-Syndrom (22q11.2-Deletionssyndrom) hat, fehlt einer Kopie des Chromosoms 22 ein Segment, das schätzungsweise 30 bis 40 Gene enthält. Viele dieser Gene wurden nicht eindeutig identifiziert und sind nicht gut verstanden. Die gelöschte Region von Chromosom 22 ist als 22q11.2 bekannt.
Die Deletion von Genen auf Chromosom 22 tritt normalerweise als zufälliges Ereignis im Sperma des Vaters oder in der Eizelle der Mutter auf, oder sie kann früh während der fötalen Entwicklung auftreten. In seltenen Fällen handelt es sich bei der Deletion um eine Erbkrankheit, die von einem Elternteil an ein Kind weitergegeben wird, der ebenfalls Deletionen in Chromosom 22 aufweist, aber möglicherweise Symptome aufweist oder nicht.
Komplikationen
Ventrikelseptumdefekt

Ventrikelseptumdefekt
Ein Ventrikelseptumdefekt ist eine abnorme Öffnung (Loch) im Herzen, die sich zwischen den unteren Pumpkammern (Ventrikeln) des Herzens bildet, wie im Herzen rechts dargestellt. Dadurch können sich sauerstoffreiches und sauerstoffarmes Blut vermischen. Links ist ein normales Herz dargestellt.
Truncus arteriosus

Truncus arteriosus
Wenn Sie oder Ihr Baby einen Truncus arteriosus haben, führt anstelle von zwei getrennten Gefäßen ein großes Gefäß aus dem Herzen und zwischen den Herzkammern befindet sich ein Loch in der Wand (Ventrikelseptumdefekt). Das sauerstoffreiche Blut (rot) und das sauerstoffarme Blut (blau) vermischen sich, wodurch Blut mit unzureichender Sauerstoffversorgung (lila) für den Körper entsteht.
Fallot-Tetralogie

Fallot-Tetralogie
Die Fallot-Tetralogie ist eine Kombination aus vier angeborenen Herzfehlern. Die vier Defekte sind ein Ventrikelseptumdefekt (VSD), eine Pulmonalstenose, eine fehlplatzierte Aorta und eine verdickte rechte Ventrikelwand (rechtsventrikuläre Hypertrophie). Sie führen normalerweise zu einem Mangel an sauerstoffreichem Blut, das den Körper erreicht.
Nebenschilddrüsen

Nebenschilddrüsen
Hinter der Schilddrüse liegen die Nebenschilddrüsen. Sie produzieren Parathormon, das eine Rolle bei der Regulierung des Blutspiegels von Kalzium und Phosphor im Körper spielt.
Gaumenspalte
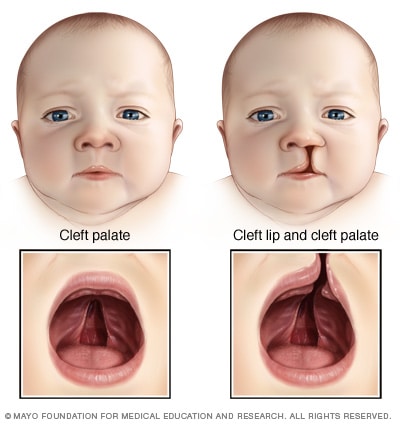
Gaumenspalte
Eine Gaumenspalte ist eine Öffnung oder Spaltung im Gaumen, die auftritt, wenn das Gewebe während der Entwicklung im Mutterleib nicht zusammenwächst. Eine Gaumenspalte beinhaltet oft eine Spaltung (Spalte) in der Oberlippe (Lippenspalte), kann aber auftreten, ohne die Lippe zu beeinträchtigen.
Teile des Immunsystems

Teile des Immunsystems
Das Lymphsystem ist Teil des körpereigenen Immunsystems, das vor Infektionen und Krankheiten schützt. Das lymphatische System umfasst Milz, Thymusdrüse, Lymphknoten und Lymphkanäle sowie Mandeln und Adenoide.
Die beim DiGeorge-Syndrom (22q11.2-Deletionssyndrom) deletierten Teile von Chromosom 22 spielen eine Rolle bei der Entwicklung einer Reihe von Körpersystemen. Infolgedessen kann die Störung mehrere Fehler während der fötalen Entwicklung verursachen. Häufige Probleme, die beim 22q11.2-Deletionssyndrom auftreten, sind:
- Herzfehler. Das 22q11.2-Deletionssyndrom verursacht häufig Herzfehler, die zu einer unzureichenden Versorgung mit sauerstoffreichem Blut führen können. Defekte können beispielsweise ein Loch zwischen den unteren Herzkammern (Ventrikelseptumdefekt) umfassen; nur ein großes Gefäß statt zwei Gefäße, das aus dem Herzen herausführt (Truncus arteriosus); oder eine Kombination von vier abnormen Herzstrukturen (Fallot-Tetralogie).
- Hypoparathyreoidismus. Die vier Nebenschilddrüsen im Nacken regulieren den Kalzium- und Phosphorspiegel im Körper. Das 22q11.2-Deletionssyndrom kann kleinere als normale Nebenschilddrüsen verursachen, die zu wenig Parathormon (PTH) absondern, was zu Hypoparathyreoidismus führt. Dieser Zustand führt zu einem niedrigen Kalziumspiegel und einem hohen Phosphorspiegel im Blut.
- Dysfunktion der Thymusdrüse. In der Thymusdrüse, die sich unter dem Brustbein befindet, reifen T-Zellen – eine Art weißer Blutkörperchen – heran. Reife T-Zellen werden benötigt, um Infektionen zu bekämpfen. Bei Kindern mit 22q11.2-Deletionssyndrom kann die Thymusdrüse klein sein oder fehlen, was zu einer schwachen Immunfunktion und häufigen, schweren Infektionen führt.
- Gaumenspalte. Eine häufige Erkrankung des 22q11.2-Deletionssyndroms ist eine Gaumenspalte – eine Öffnung (Spalte) im Gaumen (Gaumen) – mit oder ohne Lippenspalte. Andere, weniger sichtbare Anomalien des Gaumens, die ebenfalls vorhanden sein können, können das Schlucken erschweren oder bestimmte Laute beim Sprechen erzeugen.
- Ausgeprägte Gesichtszüge. Bei einigen Menschen mit 22q11.2-Deletionssyndrom können eine Reihe besonderer Gesichtszüge vorhanden sein. Dazu können kleine, tief angesetzte Ohren, kurze Augenöffnungen (Lidspalten), geschlossene Augen, ein relativ langes Gesicht, eine vergrößerte Nasenspitze (bauchig) oder eine kurze oder abgeflachte Furche in der Oberlippe gehören.
- Lern-, Verhaltens- und psychische Gesundheitsprobleme. Die Deletion von 22q11.2 kann Probleme mit der Entwicklung und Funktion des Gehirns verursachen, was zu Lern-, sozialen, Entwicklungs- oder Verhaltensproblemen führen kann. Verzögerungen in der Sprachentwicklung von Kleinkindern und Lernschwierigkeiten sind häufig. Manche Kinder entwickeln eine Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS) oder eine Autismus-Spektrum-Störung. Im späteren Leben steigt das Risiko für Depressionen, Angststörungen und andere psychische Störungen.
- Autoimmunerkrankungen. Menschen, die als Kinder aufgrund einer kleinen oder fehlenden Thymusdrüse eine schlechte Immunfunktion hatten, können auch ein erhöhtes Risiko für Autoimmunerkrankungen wie rheumatoide Arthritis oder Morbus Basedow haben.
- Andere Probleme. Eine große Anzahl von Erkrankungen kann mit dem 22q11.2-Deletionssyndrom in Verbindung gebracht werden, wie z. B. Hörbehinderung, schlechtes Sehvermögen, Atemprobleme, schlechte Nierenfunktion und relativ kleine Statur für die eigene Familie.
Verhütung
In einigen Fällen kann das DiGeorge-Syndrom (22q11.2-Deletionssyndrom) von einem betroffenen Elternteil an ein Kind weitergegeben werden. Wenn Sie sich Sorgen über eine Familienanamnese mit 22q11.2-Deletionssyndrom machen oder wenn Sie bereits ein Kind mit dem Syndrom haben, sollten Sie einen auf genetische Störungen spezialisierten Arzt (Genetiker) oder einen genetischen Berater um Hilfe bei der Planung konsultieren zukünftige Schwangerschaften.
Quellen:
- Nationalbibliothek für Medizin. 22q11.2-Deletionssyndrom. Genetik-Home-Referenz. https://ghr.nlm.nih.gov/condition/22q112-deletion-syndrome#genes. Abgerufen am 25. Mai 2017.
- 22q11.2-Deletionsstörungen (DiGeorge-Syndrom und Velokardiofaziales Syndrom). American Heart Association. http://www.heart.org/HEARTORG/Conditions/CongenitalHeartDefects/AboutCongenitalHeartDefects/22q112-Deletion-Disorders-DiGeorge-Syndrome-and-Velocardiofacial-Syndrome_UCM_309017_Article.jsp#.WSc5wWd1rRE. Abgerufen am 25. Mai 2017.
- DiGeorge-Syndrom (DGS). American Academy of Allergy Asthma & Immunology. http://www.aaaai.org/conditions-and-treatments/primary-immunodeficiency-disease/digeorge-syndrome. Abgerufen am 25. Mai 2017.
- DiGeorge-Syndrom. Merck Manual Professional-Version. https://www.merckmanuals.com/professional/immunology-allergic-disorders/immunodeficiency-disorders/digeorge-syndrome. Abgerufen am 25. Mai 2017.
- Hofstetter AM, et al. Verwendung und Sicherheit von Lebendimpfstoffen beim DiGeorge-Syndrom. Pädiatrie. 2014;133:e946.
- Chromosom 22q11.2-Deletionssyndrom. Nationale Organisation für seltene Erkrankungen. https://rarediseases.org/rare-diseases/chromosome-22q11-2-deletion-syndrome/. Abgerufen am 25. Mai 2017.
- Seroogie CM. DiGeorge (22q11.2-Deletion)-Syndrom: Epidemiologie und Pathogenese. https://www.uptodate.com/contents/search. Abgerufen am 25. Mai 2017.
- Seroogie CM. DiGeorge (22q11.2-Deletion)-Syndrom: Klinische Merkmale und Diagnose. https://www.uptodate.com/contents/search. Abgerufen am 25. Mai 2017.
- Seroogie CM. DiGeorge (22q11.2-Deletion)-Syndrom: Management und Prognose. https://www.uptodate.com/contents/search. Abgerufen am 25. Mai 2017.
- Fragen Sie MayoExpert. 22q11.2-Deletionssyndrom. Rochester, Minnesota: Mayo Foundation for Medical Education and Research; 2016. Zugriff am 10. Mai 2017.
- Babovic-Vuksanovic D (Gutachten). Mayo Clinic, Rochester, Minnesota, 6. Juli 2017.