 Suche
Suche
 Mein Konto
Mein Konto
Характеристики на рака: нови измерения

Предговор Отличителните белези на концептуализацията на рака е евристичен инструмент за дестилиране на огромната сложност на раковите фенотипове и генотипове в предварителен набор от основни принципи. С напредването на знанията за механизмите на рака, други аспекти на болестта се появиха като потенциални уточнения. Това повдига перспективата, че фенотипната пластичност и нарушената диференциация са отделна характерна способност и че немутационното епигенетично препрограмиране и полиморфните микробиоми представляват характерни позволяващи свойства, които улесняват придобиването на характерни способности. Освен това, стареещи клетки от различен произход могат да бъдат добавени към списъка на функционално важни типове клетки в туморната микросреда. Което означава, че ракът е страшен в...

Характеристики на рака: нови измерения
Предговор
Отличителните белези на концептуализацията на рака е евристичен инструмент за дестилиране на огромната сложност на раковите фенотипове и генотипове в предварителен набор от основни принципи. С напредването на знанията за механизмите на рака, други аспекти на болестта се появиха като потенциални уточнения. Това повдига перспективата, че фенотипната пластичност и нарушената диференциация са отделна характерна способност и че немутационното епигенетично препрограмиране и полиморфните микробиоми представляват характерни позволяващи свойства, които улесняват придобиването на характерни способности. Освен това, стареещи клетки от различен произход могат да бъдат добавени към списъка на функционално важни типове клетки в туморната микросреда.
Значение
Ракът е плашещ с широчината и обхвата на своето разнообразие, което включва генетика, клетъчна и тъканна биология, патология и отговор на терапията. Все по-мощните експериментални и изчислителни инструменти и технологии предоставят лавина от „големи данни“ за безбройните болестни прояви, които ракът обхваща. Интегративната концепция, въплътена в отличителните белези на рака, помага да се дестилира тази сложност във все по-логична наука и предварителните нови измерения, представени в тази перспектива, могат да добавят стойност към това усилие за по-добро разбиране на механизмите на канцерогенезата и злокачествената прогресия и прилагане на това знание към медицината на рака.
въведение
Отличителните белези на рака са предложени като набор от функционални способности, които човешките клетки придобиват, докато преминават от нормално състояние към състояния на неопластичен растеж, по-специално способности, които са критични за способността им да образуват злокачествени тумори. В тези статии ( 1, 2 ), Боб Уайнбърг и аз изброихме това, което си представихме като общи черти, които обединяват всички видове ракови клетки на ниво клетъчен фенотип. Намерението беше да се осигури концептуална рамка, която би позволила сложните фенотипове на различни типове и варианти на човешки тумори да бъдат рационализирани във връзка с общ набор от основни клетъчни параметри. Първоначално предвидихме допълващото включване на шест различни възможности на марката и по-късно разширихме този брой до осем.
Тази формулировка е повлияна от признанието, че човешките ракови заболявания се развиват като продукти на многоетапни процеси и че придобиването на тези функционални способности може да се припише по някакъв начин на отделните стъпки на туморната патогенеза. Разнообразието от злокачествена патогенеза, обхващаща множество видове тумори и нарастващо множество от подтипове, включва различни аберации (и по този начин придобити способности и свойства), които са резултат от тъканно-специфични бариери, които задължително се заобикалят по време на определени пътища на туморогенеза. Въпреки че признаваме, че такива специализирани механизми могат да бъдат полезни, ние ограничихме обозначаването на отличителните белези до параметри, които имат широко въздействие върху целия спектър от човешки ракови заболявания.
Осемте отличителни белега понастоящем включват (Фиг.1, вляво) придобитите способности за поддържане на пролиферативна сигнализация, избягване на супресори на растежа, устойчивост на клетъчна смърт, активиране на репликативно безсмъртие, индуциране/достъп до съдове, активиране на инвазия и метастази, препрограмиране на клетъчния метаболизъм и избягване на разрушаването на имунната система. В най-новата разработка на тази концепция (2), дерегулацията на клетъчния метаболизъм и избягването на разрушаването на имунната система бяха демаркирани като „нововъзникващи отличителни белези“, но сега, единадесет години по-късно, е очевидно, че подобно на оригиналните шест, те могат да се считат за основни отличителни белези на рака и са включени като такива в настоящия разказ (фиг. 1, вляво).
Фигура 1
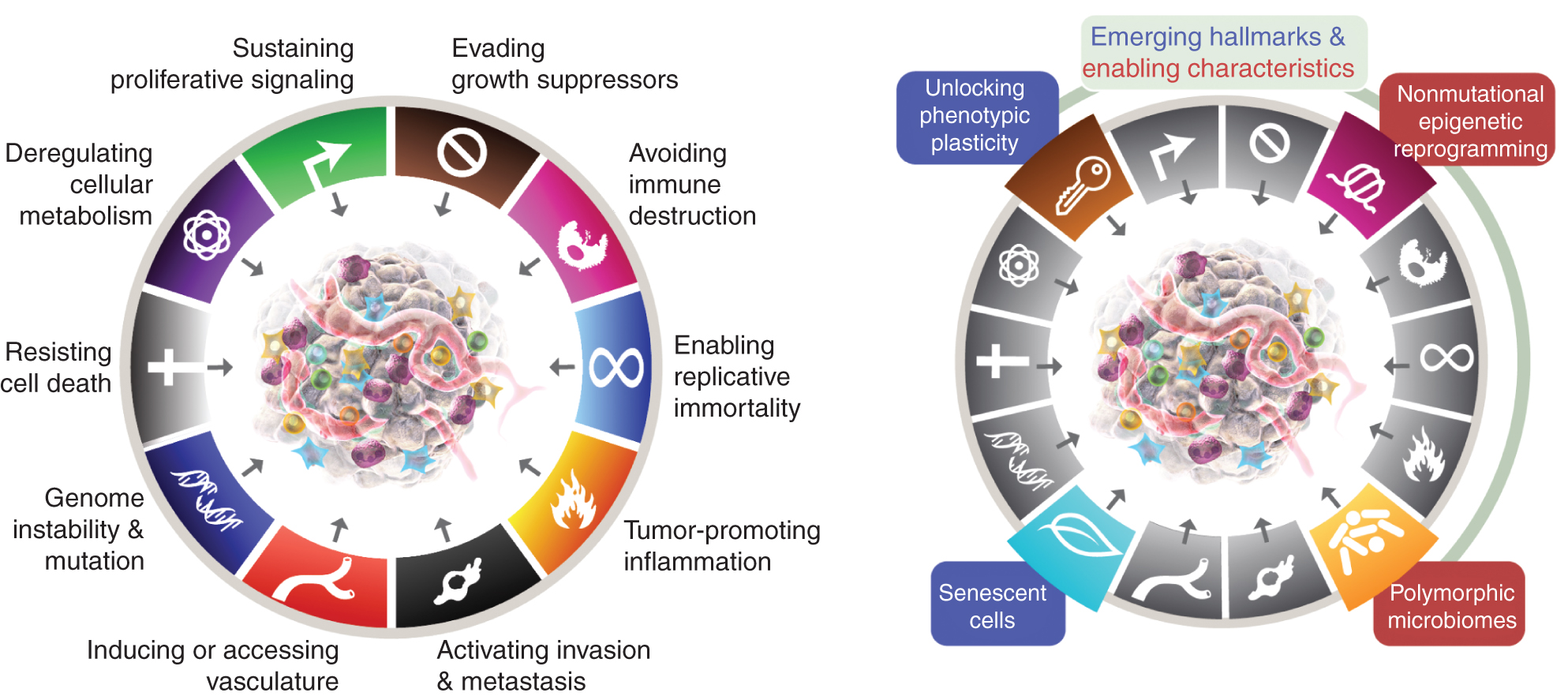
Отличителните белези на Рака в момента въплъщават осем отличителни способности и две поддържащи качества. В допълнение към шестте придобити способности – отличителни белези на рака – предложени през 2000 г. (1), двата предварителни „възникващи отличителни белези“, въведени през 2011 г. (2) – клетъчна енергетика (сега по-често наричана „препрограмиране на клетъчния метаболизъм“) и „избягване на имунна деструкция“ – са били достатъчно валидирани, за да се считат за част от основния набор.
Като се има предвид нарастващото признание, че туморите могат да бъдат адекватно васкуларизирани, или чрез включване на ангиогенеза, или чрез кооптиране на нормална тъканна васкулатура (128), този отличителен белег също е по-широко дефиниран като способността за индуциране или по друг начин достъп до васкулатура, поддържаща растежа на тумора предимно чрез инвазия и метастази.
Продължението от 2011 г. също включва „възпаление, насърчаващо тумора“ като втора благоприятна черта, допълваща всеобхватната „нестабилност и мутация на генома“, които заедно са фундаментално включени в активирането на осемте характерни (функционални) способности, необходими за растежа и прогресията на тумора. Вярно е, че този преглед включва допълнителни предложени нови отличителни белези и активиращи характеристики, включително „отключване на фенотипна пластичност“, „немутационно епигенетично препрограмиране“, „полиморфни микробиоми“ и „стареещи клетки“. Търговските марки на графиката на рака са възприети от Hanahan и Weinberg (2).
Както отбелязахме по това време, тези отличителни характеристики сами по себе си не могат да отговорят на сложността на патогенезата на рака, т.е. точните молекулярни и клетъчни механизми, които позволяват на развиващите се пренеопластични клетки да развият и придобият тези анормални фенотипни способности по време на туморогенезата и злокачествената прогресия.
Съответно, ние добавихме друга концепция към дискусията, представена като „позволяващи характеристики“, последствия от анормалното състояние на неоплазмата, които осигуряват средствата, чрез които раковите клетки и туморите могат да придобият тези функционални характеристики. Като такива, активиращите свойства се отразяват в молекулярни и клетъчни механизми, чрез които се придобиват отличителни белези, а не в самите горни осем умения. Тези два процеса на активиране са нестабилност на генома и стимулиращо тумор възпаление.
Освен това разбрахме, че туморната микросреда (TME), дефинирана тук като съставена от хетерогенни и интерактивни популации от ракови клетки и ракови стволови клетки, заедно с разнообразие от набрани типове стромални клетки - трансформираният паренхим и свързаната строма - сега е широко оценена, че играе съществена роля в туморогенезата и злокачествената прогресия.
Като се има предвид продължаващият интерес към тези формулировки и продължаващото ни намерение да насърчим текущата дискусия и усъвършенстване на схемата на Hallmarks, е уместно да разгледаме един често задаван въпрос: Има ли допълнителни характеристики на този концептуален модел, които могат да бъдат включени, като се вземе предвид необходимостта да се гарантира това? че те са широко приложими в целия спектър от човешки рак? Съответно, представям няколко потенциални нови отличителни белези и позволяващи функции, които биха могли своевременно да бъдат интегрирани като основни компоненти на отличителните белези на концептуализацията на рака.
Тези параметри са „отключване на фенотипна пластичност“, „немутационно епигенетично препрограмиране“, „полиморфни микробиоми“ и „стареещи клетки“ (фиг. 1, вдясно). Важно е, че примерите, представени в подкрепа на тези тези, са илюстративни, но в никакъв случай не са изчерпателни, тъй като има нарастващ и все по-убедителен обем от публикувани доказателства в подкрепа на всяка винетка.
Подслушване на фенотипна пластичност
По време на органогенезата, развитието, определянето и организирането на клетките в тъкани за изпълнение на хомеостатични функции е придружено от крайна диференциация, като прогениторните клетки спират да растат, понякога необратимо, тъй като тези процеси достигат кулминацията. Като такъв, крайният резултат от клетъчната диференциация в повечето случаи е антипролиферативен, образувайки ясна бариера за продължаване на пролиферацията, необходима за неоплазия.
Има все повече доказателства, че отключването на нормално ограничения капацитет за фенотипна пластичност за заобикаляне или избягване на състоянието на терминална диференциация е критичен компонент на патогенезата на рака (3). Тази пластичност може да действа в няколко проявления (фиг. 2). По този начин зараждащите се ракови клетки, които произхождат от нормална клетка, която е еволюирала по пътя, който се приближава или приема напълно диференцирано състояние, могат да обърнат курса чрез дедиференциране обратно към състояния, подобни на прогениторни клетки.
Обратно, неопластични клетки, възникващи от прогениторна клетка, предназначена да следва път, водещ до крайна диференциация, могат да съкратят процеса и да поддържат разширяващите се ракови клетки в частично диференцирано състояние, подобно на прогенитор. Алтернативно, може да възникне трансдиференциация, при която клетките, първоначално ангажирани с един път на диференциация, преминават към напълно различна програма за развитие и по този начин придобиват тъканно-специфични характеристики, които не са били предварително определени от техните нормални клетки на произход.
Следните примери подкрепят аргумента, че различните форми на клетъчна пластичност разкриват фенотипна пластичност. Отляво, фенотипната пластичност е вероятно придобита характерна способност, която позволява различни смущения на клетъчната диференциация, включително (i) дедиференциация от зрели до прогениторни състояния, (ii) застой (терминална) диференциация от прогениторни клетъчни състояния и (iii) трансдиференциация в други клетъчни линии. Три видни режима на нарушена диференциация, които са неразделна част от патогенезата на рака, са показани вдясно.
Чрез диференциално изкривяване на нормалната диференциация на прогениторни клетки в зрели клетки в линии на развитие, туморогенезата и злокачествената прогресия, произтичащи от клетките на произход в такива пътища, се улесняват. Търговските марки на графиката на рака са възприети от Hanahan и Weinberg (2).
Фигура 2

Дедиференциация
Карциногенезата на дебелото черво е пример за нарушена диференциация, тъй като има телеологична необходимост за зараждащите се ракови клетки да избягат от конвейера на терминална диференциация и ексфолиация, което по принцип може да се случи чрез дедиференциация на епителните клетки на дебелото черво, които все още не са окончателно диференцирани или чрез застой на диференциацията на прогениторни/стволови клетки в криптите, които пораждат тези диференциращи клетки. Както диференцираните клетки, така и стволовите клетки са замесени като клетки на произход за рак на дебелото черво (4 – 6).
Два транскрипционни фактора на развитието (TF), хомеобокс протеинът HOXA5 и SMAD4, последният участващ в BMP сигнализирането, са силно експресирани в диференциращите епителни клетки на дебелото черво и обикновено се губят в напреднали карциноми на дебелото черво, които характерно експресират маркери на стволови и прогениторни клетки. Функционални смущения в миши модели са показали, че принудителната експресия на HOXA5 в клетките на рак на дебелото черво възстановява маркерите за диференциация, потиска фенотипите на стволовите клетки и уврежда инвазията и метастазите, осигурявайки обосновка за характерната му регулация надолу (7, 8).
За разлика от това, SMAD4 налага както диференциация, така и потискане на пролиферацията, задвижвана от онкогенно WNT сигнализиране, което се разкрива чрез инженерната загуба на експресия на SMAD4, предоставяйки обяснение за неговата загуба на експресия, за да позволи дедиференциация и впоследствие хиперпролиферация, управлявана от WNT (5).
Трябва да се отбележи, че загубата на тези два „супресора на диференциация“ с получената дедиференциация е свързана с придобиването на други способности за отличителен белег, както и други регулатори, предизвикващи отличителен белег, усложнявайки стриктното определение на този временен отличителен белег като разделим и независим.
Друга линия от доказателства се отнася до потиснатата експресия на MITF главния регулатор на диференциацията на меланоцитите, който изглежда участва в генезиса на агресивни форми на злокачествен меланом. Загубата на този TF в развитието е свързана с реактивиране на прогениторни гени на нервния гребен и понижаване на регулацията на гени, които характеризират напълно диференцираните меланоцити. Повторната поява на гените на нервния гребен показва, че тези клетки се връщат в прогениторното състояние, от което меланоцитите възникват в развитието.
Освен това, проучване за проследяване на линията на индуцирани от BRAF меланоми установи зрели пигментирани меланоцити като клетки на произход, които претърпяват дедиференциация по време на хода на туморогенезата (9). Трябва да се отбележи, че мутантният BRAF онкоген, открит в повече от половината кожни меланоми, индуцира хиперпролиферация, която предшества и следователно може механично да бъде отделена от последващата дедиференциация, която възниква от понижаване на MITF.
Друго проучване функционално включва регулация на развитието на TF ATF2, чиято характерна експресия в миши и човешки меланоми индиректно потиска MITF1, съпътстваща злокачествена прогресия на последващо дедиференцираните меланомни клетки (10). Обратно, експресията в меланоми на мутантни форми на ATF2, които не могат да потиснат MITF, води до добре диференцирани меланоми (11).
Освен това, скорошно проучване (12) свързва дедиференциацията на линията със злокачествена прогресия на неоплазми на панкреатични островни клетки до предразположени към метастази карциноми; тези невроендокринни клетки и производни тумори възникват от линия на развитие, различна от тази, която генерира много по-голям брой съседни клетки, които образуват екзокринната и панкреаса и произтичащите от това дуктални аденокарциноми.
Забележително е, че многостепенният път на диференциация от островни прогениторни клетки до зрели β-клетки е подробно характеризиран (13). Сравнителното профилиране на транскриптоми показва, че аденомоподобните островни тумори са най-сходни с незрели, но диференцирани инсулин-продуциращи β-клетки, докато инвазивните карциноми са най-сходни с ембрионалните островни клетъчни прекурсори. Прогресията до слабо диференцирани карциноми включва начална стъпка на дедиференциация, която първоначално не включва повишена пролиферация или намалена апоптоза в сравнение с добре диференцираните аденоми, като и двете са склонни да се появят по-късно.
По този начин, дискретната стъпка на дедиференциация не се задвижва от наблюдавани промени в характерните характеристики на продължителна пролиферация и резистентност към апоптоза. По-скоро регулирането нагоре на miPHK, замесено преди това в уточняването на прогениторното състояние на острова, е такова, което се регулира надолу по време на крайната диференциация на β-клетки, 12).
Блокирана диференциация
Докато горните примери илюстрират как потискането на експресията на фактор на диференциация може да улесни туморогенезата, като позволи на по-добре диференцираните клетки да се дедиференцират в прогенитори, в други случаи ненапълно диференцираните прогениторни клетки могат да претърпят регулаторни промени, които активно блокират по-нататъшното им прогресиране в напълно диференцирани, обикновено непролиферативни състояния.
Отдавна е документирано, че острата промиелоцитна левкемия (APL) е резултат от хромозомна транслокация, която слива локуса на PML с гена, кодиращ α ядрения рецептор на ретиновата киселина (RARα). Миелоидните прогениторни клетки, носещи такива транслокации, очевидно не са в състояние да продължат обичайната си крайна диференциация в гранулоцити, което води до клетки, уловени в пролиферативен, промиелоцит-подобен прогениторен стадий (14).
Доказателство за концепцията за тази схема идва от лечението на култивирани APL клетки, миши модели на заболяването и засегнати пациенти с ретиноева киселина, лиганда на RARα; Това терапевтично лечение кара неопластичните APL клетки да се диференцират в очевидно зрели, непролифериращи гранулоцити, като по този начин се прекъсва тяхната прогресивна пролиферативна експанзия (14-16).
Вариация на тази тема се отнася до друга форма на остра миелоидна левкемия, тази, носеща t(8;21) транслокация, която произвежда слетия протеин AML1-ETO. Този протеин сам по себе си може да трансформира миелоидните прогенитори, поне отчасти чрез блокиране на тяхната диференциация. Терапевтичната интервенция при миши модели и пациенти с фармакологичен инхибитор на хроматин-модифицираща хистон деацетилаза (HDAC) кара клетките на миелоидна левкемия да възобновят диференциацията в клетки с по-зряла морфология на миелоидните клетки. Придружаваща тази реакция е намаляване на пролиферативния капацитет, като по този начин се нарушава прогресията на тази левкемия ( 17 , 18 ).
Трети пример при меланома включва развитие на TF, SOX10, което обикновено се регулира надолу по време на диференциацията на меланоцитите. Проучванията за усилване и загуба на функция в модел на рибка зебра на BRAF-индуцирани меланоми показват, че необичайно поддържаната експресия на SOX10 блокира диференциацията на невралните прогениторни клетки в меланоцити, което позволява образуването на BRAF-задвижвани меланоми (19).
Други примери за модулатори на диференциация включват метаболита алфа-кетоглутарат (αKG), необходим кофактор за редица модифициращи хроматин ензими, за които е доказано, че участват в стимулирането на определени диференцирани клетъчни състояния. При рак на панкреаса, туморният супресор p53 стимулира производството на αKG и поддържането на по-диференцирано клетъчно състояние, докато прототипната загуба на функцията на p53 води до намаляване на нивата на αKG и последваща дедиференциация, която е свързана със злокачествена прогресия (20).
При една форма на рак на черния дроб, мутацията на ген за изоцитрат дехидрогеназа (IDH1/2) не води до производството на индуциращ диференциацията αKG, а по-скоро до свързан „онкометаболит“, D-2-хидроксиглутерат (D2HG), за който е доказано, че блокира хепатоцитната диференциация на чернодробни прогениторни клетки чрез D2HG-медиирана репресия на главен регулатор на хепатоцитна диференциация и покой, HNF4a.
D2HG-медиираната супресия на HNF4a функцията предизвиква пролиферативна експанзия на хепатоцитни прогениторни клетки в черния дроб, които стават податливи на онкогенна трансформация при последващо мутационно активиране на KRAS онкогена, което води до злокачествена прогресия до холангиокарцином на черния дроб (21). Мутантът IDH1/2 и неговият онкометаболит D2HG също функционират в различни видове миелоидни и други солидни тумори, където D2HG инхибира αKG-зависими диоксигенази, необходими за събития на метилиране на хистон и ДНК, които медиират промени в структурата на хроматина по време на диференциация на линията на развитие, като по този начин замразяват появата на ракови клетки в прогениторно състояние (22, 23).
Допълнителна свързана концепция е „заобиколена диференциация“, при която частично или недиференцирани прогениторни/стволови клетки излизат от клетъчния цикъл и лежат латентни в защитни ниши, с потенциала да възобновят пролиферативната експанзия (24), въпреки че все още имат селективен натиск да нарушат програмираната си диференциация по един или друг начин.
Трансдиференциация
Концепцията за трансдиференциация отдавна е призната от патолозите под формата на тъканна метаплазия, при която клетки от определен диференциран фенотип значително променят своята морфология, за да станат ясно разпознаваеми като елементи на друга тъкан, виден пример за което е хранопроводът на Барет, където хроничното възпаление на стратифицирания сквамозен епител на хранопровода индуцира трансдиференциация в прост колонен епител, характерен за червата, като по този начин улеснява последващото развитие на аденокарциноми, а не плоскоклетъчни карциноми, очаквани от този плоскоклетъчен епител (3).
Сега молекулярните детерминанти разкриват механизми на трансдиференциация при различни ракови заболявания, както за случаите, когато грубата тъканна метаплазия е очевидна, така и за други, където тя е малко по-фина, както илюстрират следващите примери.
Информативен случай за трансдиференциация като дискретно събитие в туморогенезата се отнася до дуктален аденокарцином на панкреаса (PDAC), при който една от участващите клетки на произход, ацинарната клетка на панкреаса, може да се трансдиференцира във фенотип на дуктален клетъчен по време на инициирането на неопластично развитие. Два TFs—PTF1a и MIST1—контролират спецификацията и поддържането на състоянието на диференцираните ацинарни клетки на панкреаса чрез тяхната експресия в контекста на самоподдържащи се регулаторни вериги „подаване напред” (25).
И двата TFs често се регулират надолу по време на неопластично развитие и злокачествена прогресия на човешки и миши PDAC. Функционални генетични изследвания при мишки и култивирани човешки PDAC клетки показват, че експериментално принудителното експресиране на PTF1a уврежда индуцираната от KRAS трансдиференциация и пролиферация и може също така да принуди повторното диференциране на вече неопластични клетки в неподвижен ацинарен клетъчен фенотип (26).
Обратно, потискането на експресията на PTF1a задейства метаплазия от ацинар към канал, а именно трансдиференциация, и по този начин сенсибилизира подобните на канал клетки към онкогенна KRAS трансформация, ускорявайки последващото развитие на инвазивен PDAC (27). По същия начин, принудителната експресия на MIST1 в KRAS-експресиращ панкреас също блокира трансдиференциацията и уврежда инициирането на панкреатична туморогенеза, което иначе се улеснява от образуването на премалигнени каналоподобни (PanIN) лезии, докато генетичната делеция на MIST1 засилва тяхното образуване и инициирането на KRAS-управлявана неопластична прогресия (28).
Загубата на експресия на PTF1 или MIST1 по време на туморогенезата е свързана с повишена експресия на друг регулаторен TF на развитието, SOX9, който обикновено е ефективен при спецификацията на дукталните клетки (27, 28). Принудително повишаване на регулацията на SOX9, като по този начин се избягва необходимостта от понижаване на PTF1a и MIST1 също е доказано, че стимулира трансдиференциацията на ацинарни клетки в дуктален клетъчен фенотип, чувствителен към KRAS-индуцирана неоплазия (29), намесвайки SOX9 като ключов функционален ефектор на тяхното понижаване в генезиса на човешки PDAC.
По този начин, три TF, които регулират диференциацията на панкреаса, могат да бъдат променени по различни начини, за да индуцират трансдиференцирано състояние, което в контекста на мутационно активиране на KRAS улеснява онкогенната трансформация и инициирането на туморогенеза и злокачествена прогресия.
Допълнителни членове на фамилията SOX от свързани с хроматин регулаторни фактори са, от една страна, до голяма степен свързани както със спецификацията на клетъчната съдба, така и с превключването на линията в развитието (30), а от друга страна, с няколко фенотипа, свързани с тумора (31). Друг важен пример за SOX-медиирана трансдиференциация включва механизъм на терапевтична резистентност при рак на простатата.
В този случай загубата на RB и p53 туморни супресори - чието отсъствие е характерно за невроендокринните тумори - в отговор на антиандрогенна терапия е необходима, но не е достатъчна за често наблюдаваната трансформация на добре диференцирани ракови клетки на простатата в карциномни клетки, които са нахлули в линията на диференциация с молекулярни и хистологични характеристики на невроендокринни клетки, които по-специално не експресират андрогенния рецептор. В допълнение към загубата на RB и p53, придобитата резистентност към антиандрогенна терапия изисква повишена експресия на SOX2, регулаторен ген за развитието, за който е доказано, че помага за индуциране на трансдиференциация на реагиращите на терапия аденокарциномни клетки в производни, които са в невроендокринно клетъчно състояние, рефрактерно на терапия (32).
Трети пример също показва трансдиференциация като стратегия, използвана от карциномни клетки, за да се избегне елиминирането чрез специфична за линията терапия, в този случай с базалноклетъчни карциноми (BCC) на кожата, лекувани с фармакологичен инхибитор на Hedgehog-Smoothened (HH/SMO) онкогенен път, за който е известно, че стимулира неопластичния растеж на тези клетки (33).
Устойчивите на лекарства ракови клетки преминават към свързан с развитието, но различен клетъчен тип чрез широки епигенетични промени в специфични хроматинови домени и променена достъпност на два суперенхансера. Новопридобито фенотипно състояние на BCC клетки им позволява да поддържат експресията на онкогенния WNT сигнален път, който от своя страна осигурява независимост от медикаментозно потиснатия HH/SMO сигнален път (34).
Както се очаква от тази трансдиференциация, транскриптомът на раковите клетки се измества от генен подпис, отразяващ включената клетка на произход на BCCs, а именно стволовите клетки на изпъкналостта на космения фоликул, към подпис, показателен за базалните стволови клетки, населяващи BCC интерфоликуларния епидермис. Такава трансдиференциация, позволяваща лекарствена резистентност, все повече се документира при различни форми на рак (35).
Пластичността на линията на развитие също изглежда преобладаваща в основните подтипове на белодробен карцином, т.е. при невроендокринни карциноми [дребноклетъчен рак на белия дроб (SCLC)] и аденокарциноми + плоскоклетъчен карцином [колективен недребноклетъчен рак на белия дроб (NSCLC)]. Секвенирането на едноклетъчна РНК разкри забележително динамично и хетерогенно превръщане между тези подтипове, както и изразени вариации в тях, по време на етапите на белодробна туморогенеза, последваща злокачествена прогресия и отговор на терапия (36-38).
Следователно, вместо простото концептуализиране на чисто клоново преминаване от една линия към друга, тези изследвания рисуват много по-сложна картина на динамично взаимопреобразуващи се субпопулации от ракови клетки, които проявяват характеристики на множество линии на развитие и етапи на диференциация, отрезвяващо прозрение в това отношение за базирано на линията терапевтично насочване на човешки рак на белия дроб. Регулаторните детерминанти на тази динамична фенотипна пластичност започват да се идентифицират (37, 39, 40).
Резюме
Трите класа механизми, описани по-горе, подчертават селективни регулатори на клетъчната пластичност, които са – поне частично – отделими от основните онкогенни драйвери и други отличителни способности. Отвъд тези примери има значителен набор от доказателства, свързващи много форми на рак с нарушена диференциация, която е придружена от придобиване на сигнатури на транскриптоми и други фенотипове - например хистологична морфология - които са свързани с прогениторни или етапи на стволови клетки, наблюдавани в съответните нормални тъкани. произход или в други по-отдалечени клетъчни типове и линии (41 – 43).
Като такива, тези три подкласа на фенотипна пластичност - дедиференциране на зрели клетки обратно към прогениторни състояния, забавена диференциация за замразяване на развиващи се клетки в прогениторни/стволови клетки и трансдиференциация към алтернативни клетъчни линии - изглеждат ефективни при няколко типа рак по време на първична туморогенеза, злокачествена прогресия и/или отговор на терапия.
Има обаче две концептуални съображения. Първо, дедиференциацията и застойната диференциация вероятно са преплетени, тъй като те са неразличими в много видове тумори, в които клетката на произход - диференцирана клетка или прогениторна/стволова клетка - е или неизвестна, или алтернативно включена. Второ, придобиването или поддържането на фенотипове на прогениторни клетки и загубата на диференцирани характеристики е в повечето случаи неточно отражение на нормалния етап на развитие, потапяйки се в среда на други характерни промени в раковата клетка, които не присъстват в естествено развиващите се клетки.
Освен това, друга форма на фенотипна пластичност включва клетъчно стареене, обсъдено по-общо по-долу, при което раковите клетки, предизвикани да претърпят привидно необратимо стареене, вместо това могат да избягат и да продължат пролиферативната експанзия (44). И накрая, както при други отличителни способности, клетъчната пластичност не е ново изобретение или аберация на раковите клетки, а по-скоро разваляне на латентни, но активируеми способности, които различни нормални клетки използват за поддържане на хомеостаза, възстановяване и регенерация (45).
Като цяло, тези илюстративни примери насърчават обмислянето, че отключването на клетъчната пластичност, за да се даде възможност на различни форми на нарушена диференциация, представлява отделна отличителна способност, която се различава по регулация и клетъчен фенотип от добре валидираните основни отличителни белези на рака (фиг. 2).
Епигенетично препрограмиране без мутация
Благоприятното свойство на нестабилността и мутацията на генома (ДНК) е основен компонент на развитието и патогенезата на рака. Понастоящем няколко международни консорциума каталогизират мутации в целия геном на човешки ракови клетки, в почти всеки тип човешки рак, на различни етапи на злокачествена прогресия, включително метастатични лезии, и по време на развитието на резистентност към адаптивна терапия. Един резултат е вече широко разпространеното признание, че мутациите в гените, които организират, модулират и поддържат архитектурата на хроматина и по този начин регулират генната експресия в световен мащаб, все повече се откриват и функционално се свързват с белези на рак (46-48).
Освен това има аргументи за друга привидно независима форма на препрограмиране на генома, която включва чисто епигенетично регулирани промени в генната експресия, такава, която може да се нарече „немутационно епигенетично препрограмиране“ (фиг. 3). Всъщност тезата за еволюцията на рака без мутации и чисто епигенетичното програмиране на характерни ракови фенотипове беше повдигната преди почти десетилетие (49) и все повече се обсъжда (46, 50–52).
Фигура 3

Подобно на това, което се случва по време на ембриогенезата и тъканната диференциация и хомеостаза, натрупването на доказателства предполага, че инструменталните генни регулаторни вериги и мрежи в туморите могат да бъдат контролирани от множество повредени и кооптирани механизми, които са независими от нестабилността на генома и генната мутация. Търговските марки на графиката на рака са възприети от Hanahan и Weinberg (2).
Разбира се, концепцията за немутационна епигенетична регулация на генната експресия е добре установена като централен механизъм, медииращ ембрионалното развитие, диференциация и органогенеза (53-55). При възрастни, например, дългосрочната памет включва промени в генната и хистонова модификация, в структурата на хроматина и в задействането на генни експресионни превключватели, които се поддържат стабилно във времето чрез положителни и отрицателни вериги за обратна връзка (56, 57). Все повече доказателства подкрепят идеята, че аналогични епигенетични промени могат да допринесат за придобиването на характерни способности по време на развитието на тумора и злокачествената прогресия. В подкрепа на тази хипотеза по-долу са представени някои примери.
Микрооколни механизми на епигенетично препрограмиране
Ако не само чрез онкогенни мутации, как се препрограмира геномът на раковата клетка? Все повече доказателства предполагат, че анормалните физически свойства на туморната микросреда могат да причинят широки промени в епигенома, от които промени, полезни за фенотипната селекция на способностите на чертите, могат да доведат до клонално израстване на ракови клетки с подобрена годност за пролиферативна експанзия.
Обща характеристика на туморите (или областите в туморите) е хипоксията в резултат на неадекватна васкуларизация. Хипоксията, например, намалява активността на TET деметилазите, което води до значителни промени в метилома, особено хиперметилиране ( 58 ). Недостатъчната васкуларизация също е вероятно да ограничи бионаличността на критични кръвни хранителни вещества и лишаването от хранителни вещества, например, е доказано, че променя контрола на транслацията и следователно увеличава злокачествения фенотип на раковите клетки на гърдата ( 59 ).
Убедителен пример за епигенетична регулация, медиирана от хипоксия, е форма на неизменно фаталния педиатричен епендимом. Подобно на много ембрионални и педиатрични тумори, тази форма няма повтарящи се мутации, особено липса на мутации на водача в онкогени и туморни супресори. По-скоро е доказано, че анормалният растеж на тези ракови клетки се контролира от индуцирана от хипоксия генна регулаторна програма (60, 61). Забележително е, че предполагаемата клетка на произход на този рак се намира в хипоксично отделение и вероятно сенсибилизира клетките в него, за да инициира туморогенеза чрез все още неизвестни кофактори.
Друго убедително доказателство за епигенетична регулация, медиирана от микросредата, се отнася до инвазивната способност за растеж на раковите клетки. Класически пример е обратимата индукция на инвазивност на ракови клетки в краищата на много солидни тумори, оркестрирана от регулаторната програма за развитие, известна като епителен към мезенхимален преход (EMT; реф. 62–64). По-специално, наскоро беше показано, че главен регулатор на EMT, ZEB1, индуцира експресията на хистон метилтрансфераза, SETD1B, която от своя страна поддържа ZEB1 експресията в положителна обратна връзка, която поддържа (инвазивното) регулаторно състояние на EMT (65).
Предишно проучване документира по подобен начин, че индукцията на EMT чрез повишена експресия на свързан TF, SNAIL1, причинява изразени промени в хроматиновия пейзаж в резултат на индукцията на редица хроматинови модификатори, чиято активност е доказана като необходима за поддържане на фенотипното състояние (66). Освен това редица състояния и фактори, изпитвани от раковите клетки в краищата на туморите, включително хипоксия и цитокини, секретирани от стромални клетки, очевидно могат да индуцират ЕМТ и по този начин инвазивност (67, 68).
Удивителен пример за програмиране на инвазивност от микросредата, за която се предполага, че няма връзка с програмата EMT, включва автокринното активиране на невронна сигнална верига, включваща секретиран глутамат и неговия рецептор NMDAR (69, 70). Забележително е, че прототипната твърдост на много солидни тумори, въплътена в екстензивни промени в извънклетъчната матрица (ECM), която обвива клетките в тях, има дълбоки последици за инвазивните и други фенотипни свойства на раковите клетки.
В сравнение с нормалната тъкан ECM, от която възникват туморите, туморната ECM обикновено се характеризира с повишено омрежване и плътност, ензимни модификации и променен молекулен състав, които колективно организират, отчасти чрез интегринови рецептори за ECM мотиви, индуцирано от скованост сигнализиране и мрежи за генна експресия, които индуцират инвазивност и други характерни характеристики (71).
В допълнение към такива регулаторни механизми, предоставени от физическата микросреда на тумора, паракринното сигнализиране, включващо разтворими фактори, освободени в извънклетъчната среда от различните типове клетки, които населяват солидни тумори, може също да допринесе за индуцирането на няколко морфологично различни инвазивни програми за растеж (72), само една от които – наречена „мезенхимална“ – изглежда участва в гореспоменатата епигенетика регулаторен механизъм на EMT.
Епигенетична регулаторна хетерогенност
Нарастващата база от знания повишава оценката за значението на интратуморната хетерогенност при генерирането на фенотипно разнообразие, където най-подходящите клетки за пролиферативна експанзия и инвазия надминават своите братя и следователно са избрани за злокачествена прогресия. Разбира се, един аспект на тази фенотипна хетерогенност се дължи на хронична или епизодична геномна нестабилност и произтичаща от това генетична хетерогенност в клетките, които населяват тумора.
Освен това става все по-ясно, че може да съществува не-базирана на мутации епигенетична хетерогенност. Ярък пример е линкерният хистон H1.0, който е динамично експресиран и потиснат в субпопулации от ракови клетки в рамките на редица видове тумори, с последваща секвестрация или достъпност на домейни с размер на мегабаза [73]. По-специално, установено е, че популацията от ракови клетки с потиснат H1.0 проявява свойства, подобни на стъблото, повишена способност за иницииране на тумор и връзка с лоша прогноза при пациентите.
Друг пример за епигенетично регулирана пластичност е описан при човешки орални плоскоклетъчни карциноми (SCC), където раковите клетки в инвазивните граници приемат частично EMT състояние (p-EMT), което няма гореспоменатите мезенхимни TFs, но експресира други EMT-дефиниращи гени, които не се експресират в централното ядро на туморите (74).
Клетките p-EMT очевидно не представляват клонова компартментализация на мутационно променени клетки: култури от ракови клетки, получени от първичен тумор, съдържат динамични смеси както от p-EMT hi, така и от p-EMT lo клетки и когато p-EMT hi/lo клетките бяха FACS пречистени и култивирани, и двете върнати към смесени популации от p-EMT hi и p-EMT lo в рамките на 4 дни. Въпреки че паракринните сигнали от съседната строма могат да се считат за детерминистични за състоянието на p-EMT hi, стабилното присъствие и регенерацията на двете епигенетични състояния в културата доказва наличието на механизъм, присъщ на раковите клетки. Трябва да се отбележи, че това заключение се подкрепя от анализа на 198 клетъчни линии, представляващи 22 типа рак, включително SCC, където 12 стабилно хетерогенни епигенетични състояния (включително p-EMT в SCC) са открити по различен начин в моделите на клетъчната линия, както и техните свързани първични тумори (75).
Отново, хетерогенните фенотипни състояния не могат да бъдат свързани с откриваеми генетични разлики и в няколко случая е доказано, че FACS-сортирани клетки от определено състояние динамично се уравновесяват отново при културата, рекапитулирайки стабилно равновесие между хетерогенните състояния, наблюдавани в оригиналните клетъчни линии.
Освен това, технологиите за профилиране в целия геном на различни атрибути – отвъд ДНК последователността и нейните мутационни вариации – осветяват влиятелни елементи от анотацията и организацията на генома на раковите клетки, които корелират с прогнозата на пациента и, все повече, с характерните способности (76 – 78). Епигеномната хетерогенност се разкрива чрез все по-мощни технологии за профилиране на метилиране на ДНК в целия геном (79, 80), модификация на хистони (81), достъпност на хроматин (82) и пост-транскрипционна модификация и транслация на РНК (83, 84).
Предизвикателство по отношение на разглеждания тук постулат ще бъде да се определи кои епигеномни модификации в определени типове рак (i) имат регулаторно значение и (ii) са представителни за чисто немутационно препрограмиране, за разлика от мутационно задвижваната и по този начин обяснима с генома нестабилност.
Епигенетична регулация на типове стромални клетки, населяващи туморната микросреда
Като цяло, не се смята, че спомагателните клетки в туморната микросреда, които функционално допринасят за придобиването на характерни способности, страдат от генетична нестабилност и мутационно препрограмиране, за да се засилят техните стимулиращи тумора дейности; по-скоро се заключава, че тези клетки – асоциирани с рака фибробласти, вродени имунни клетки и ендотелни клетки и перицити на туморната васкулатура – са епигенетично препрограмирани при тяхното набиране от разтворими и физически фактори, които определят микросредата на солиден тумор (2, 85).
Очаква се технологиите за мулти-омично профилиране, прилагани в момента към раковите клетки, да се използват все повече за изследване на допълнителните (стромални) клетки в тумори, за да се изясни как нормалните клетки са увредени, за да поддържат функционално развитието и прогресията на тумора. Например, скорошно проучване (86) предполага, че такова препрограмиране може да включва модификации на епигенома, в допълнение към индуктивния обмен на цитокини, хемокини и растежни фактори, които променят вътреклетъчните сигнални мрежи във всички тези типове клетки:
Когато миши модели с белодробни метастази бяха третирани с комбинация от инхибитор на ДНК метилтрансфераза (5-азацитидин) и инхибитор на модификация на хистон (HDAC), беше установено, че инфилтриращите миелоидни клетки са преминали от незряло (туморно стимулиращо) прогениторно състояние в клетки, наподобяващи зрели интерстициални (туморно-антагонизиращи) макрофаги, които, за разлика от своите двойници в нелекувани тумори, не са били в състояние да поддържат типичните способности, необходими за ефективна метастатична колонизация (86). Възможно е мултиомичното профилиране и фармакологичните смущения да послужат за изясняване на препрограмираното епигенетично състояние в такива миелоидни клетки, както и други характерни спомагателни типове клетки, които населяват туморни микросреди.
Резюме
Взети заедно, тези илюстративни моментни снимки подкрепят тезата, че епигенетичното препрограмиране без мутация ще бъде прието като истинска благоприятна черта, която служи за улесняване на придобиването на характерни способности (фиг. 3), различни от нестабилността и мутацията на геномната ДНК. По-специално, може да се очаква немутационното епигенетично препрограмиране да се окаже неразделна част от позволяването на предварителната нова отличителна способност на фенотипната пластичност, обсъдена по-горе, особено като движеща сила в динамичната транскриптомна хетерогенност, която е все по-добре документирана в ТМЕ на злокачествени ракови клетки. Напредъкът на едноклетъчните многоомични технологии за профилиране ще хвърли светлина върху съответните приноси и взаимодействие между управлявана от мутация и неуправлявана от мутация епигенетична регулация в развитието на тумори по време на злокачествена прогресия и метастази.
Полиморфни микробиоми
Една широкообхватна граница в биомедицината се разгръща чрез осветяване на разнообразието и променливостта на изобилието от микроорганизми, наричани колективно микробиота, които се свързват симбиотично с бариерните тъкани на тялото, изложени на външната среда - особено епидермиса и вътрешната лигавица на стомашно-чревния тракт, както и белите дробове, гърдите и пикочно-половата система.
Все повече се признава, че екосистемите, създадени от резидентни бактерии и гъбички – микробиомите – имат дълбок ефект върху здравето и болестите (87), осъзнаване, което се дължи на способността за скрининг на популациите на микробни видове с помощта на следващо поколение секвениране и биоинформационни технологии. За рака доказателствата стават все по-убедителни, че полиморфната вариабилност в микробиомите между индивидите в популацията може да има дълбоки ефекти върху раковите фенотипове (88, 89).
Проучвания на асоциации при хора и експериментални манипулации при миши модели на рак разкриват определени микроорганизми, предимно, но не изключително бактерии, които могат да имат или защитни, или вредни ефекти върху развитието на рак, злокачествената прогресия и отговора на терапията. Това се отнася и за глобалната сложност и състав на тъканния микробиом като цяло. Докато микробиомът на червата беше пионерът на тази нова граница, няколко тъкани и органи имат свързани микробиоми, които проявяват отличителни характеристики, свързани с динамиката на популацията и разнообразието от микробни видове и подвидове.
Това нарастващо оценяване на важността на полиморфно променливите микробиоми в здравето и болестта повдига въпроса: Дали микробиомът е отделна благоприятна черта, която широко влияе, както положително, така и отрицателно, върху придобиването на отличителни способности за рак? Разглеждам тази възможност по-долу и илюстрирам доказателства за някои от видните тъканни микробиоми, замесени в чертите на рака (фиг. 4), като се започне с най-видния и очевидно най-въздействащ микробиом, този на чревния тракт.
Фигура 4
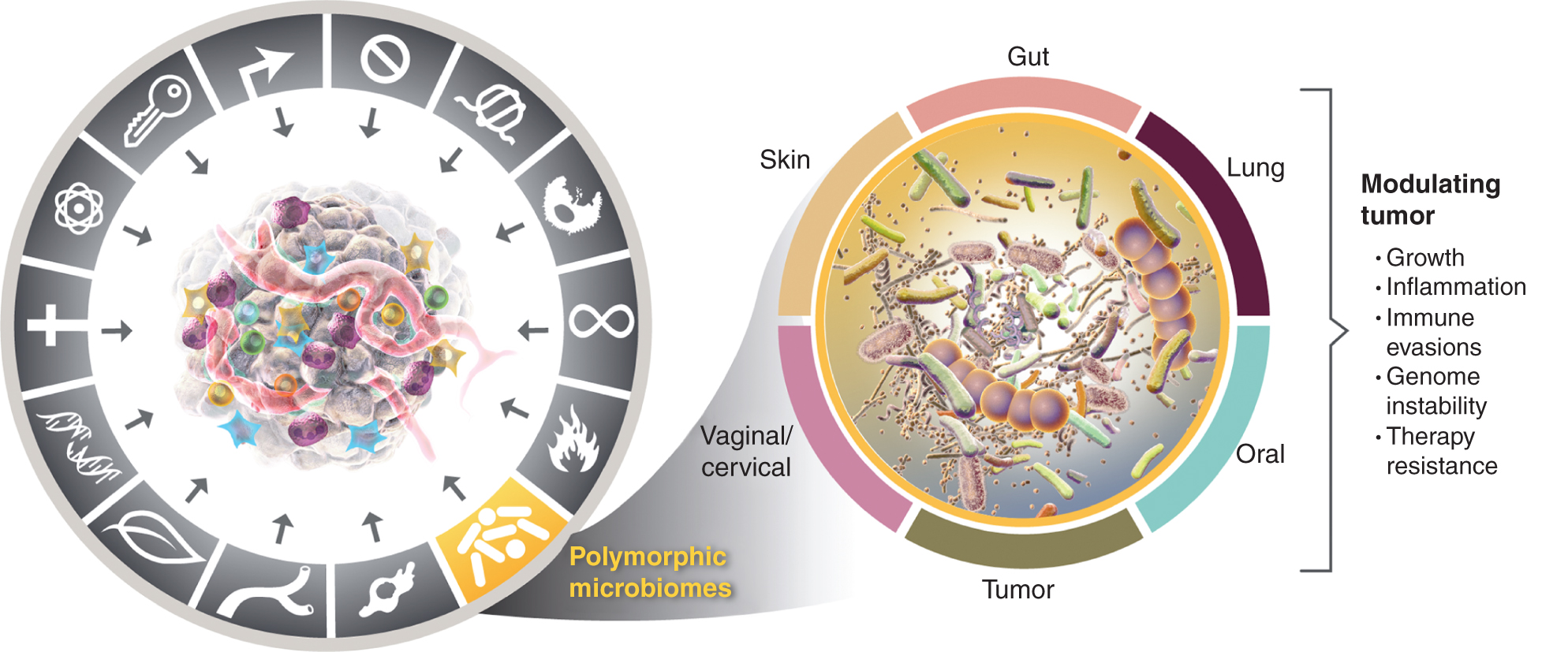
Отляво, докато активиращите свойства на стимулиращото тумор възпаление и геномната нестабилност и мутация се припокриват, има все повече основания да се заключи, че полиморфните микробиоми, разположени в един индивид в сравнение с друг в дебелото черво, в други лигавици и свързани органи или в самите тумори, могат да повлияят много от характерните способности по различни начини - или чрез индукция, или инхибиране - и следователно могат да бъдат инструментални и квазинезависими променлива в пъзела за това как ракът се развива, прогресира и расте реагира на терапията. Вярно е, че множество тъканни микробиоми участват в модулирането на туморните фенотипове. В допълнение към широко изследваната чревна микробиома, други характерни тъканни микробиоми, както и туморната микробиома участват в модулирането на придобиването - както положително, така и отрицателно - на характерните способности, представени в определени видове тумори. Търговските марки на графиката на рака са възприети от Hanahan и Weinberg (2).
Множество модулиращи ефекти на чревния микробиом
Отдавна е известно, че чревната микробиома е фундаментална за функцията на дебелото черво (дебелото черво) при разграждането и внасянето на хранителни вещества в тялото като част от метаболитната хомеостаза и че нарушаването на микробните популации - дисбиоза - в дебелото черво може да причини спектър от физиологични заболявания (87). Това включва подозрението, че чувствителността, развитието и патогенезата на рак на дебелото черво се влияят от чревния микробиом. През последните години убедителни функционални проучвания, използващи фекални трансплантации от пациенти с тумор на дебелото черво и мишки в мишки реципиенти, предразположени към развитие на рак на дебелото черво, установиха принцип: съществуват както предпазващи от рак, така и стимулиращи тумори микробиоми, включващи специфични бактериални видове, които могат да модулират появата и патогенезата на тумори на дебелото черво (90).
Механизмите, чрез които микробиотата придава тези модулиращи роли, все още се изясняват, но два общи ефекта са все по-добре установени за микробиомите, стимулиращи тумора, и в някои случаи за специфични бактериални видове, стимулиращи тумора. Първият ефект е мутагенеза на епитела на дебелото черво в резултат на производството на бактериални токсини и други молекули, които или директно увреждат ДНК, или нарушават системите, които поддържат геномната цялост или по друг начин стресират клетките, косвено засягащи верността на репликацията и възстановяването на ДНК. Типичен пример е Е. coli, която носи PKS локуса, за който е доказано, че мутагенизира човешкия геном и участва в предаването на мутации, които позволяват марката (91).
Освен това се съобщава, че бактериите се свързват с повърхността на епителните клетки на дебелото черво и произвеждат лигандни миметици, които стимулират епителната пролиферация, допринасяйки за характерната пролиферативна сигнална способност в неопластични клетки (88). Друг механизъм, чрез който специфични видове бактерии насърчават развитието на тумори, са бактериите, произвеждащи бутират, чието изобилие се увеличава при пациенти с колоректален рак (92).
Производството на метаболита бутират има сложни физиологични ефекти, включително индукция на стареене на епителни и фибробластни клетки. Миши модел на карциногенеза на дебелото черво, колонизиран с бактерии, произвеждащи бутират, развива повече тумори, отколкото мишки без такива бактерии; Връзката между индуцираното от бутират стареене и повишената туморогенеза на дебелото черво е доказана чрез използването на сенолитично лекарство, което убива стареещите клетки, нарушавайки туморния растеж (92).
Освен това, бактериално произведеният бутират има плейотропни и парадоксални ефекти върху диференцираните клетки в сравнение с недиференцираните (стволови) клетки в епитела на дебелото черво при състояния, при които чревната бариера е нарушена (дисбиоза) и бактериите са инвазивни, засягайки например клетъчната енергия и метаболизма, хистонова модификация, прогресия на клетъчния цикъл и (стимулиращ тумора) вроден имунен възпаление, което имуносупресира адаптивните имунни отговори (93).
В действителност широкото действие на полиморфните микробиоми включва модулирането на адаптивната и вродена имунна система чрез различни пътища, включително производството на „имуномодулиращи“ фактори от бактерии, които активират сензори за увреждане на епителните или резидентните имунни клетки, което води до експресията на разнообразен репертоар от хемокини и цитокини, които могат да оформят изобилието и свойствата на имунните клетки, населяващи епител на дебелото черво и подлежащата строма и дрениращи лимфни възли.
В допълнение, някои бактерии могат да нарушат както защитния биофилм, така и слузта, облицоващи епитела на дебелото черво, и да нарушат плътните връзки между епителните клетки и клетките, които колективно поддържат целостта на физическата бариера, която обикновено разделя микробиома на червата. При нахлуване в стромата бактериите могат да предизвикат както вродени, така и адаптивни имунни отговори, като предизвикат секрецията на репертоар от цитокини и хемокини. Една проява може да бъде създаването на тумор-стимулиращи или тумор-антагонизиращи имунни микросреди, които впоследствие предпазват или улесняват туморогенезата и злокачествената прогресия.
Съответно, модулирането на преплетените параметри на (i) индуцирането на (вродено) стимулиращо тумора възпаление и (ii) бягството от (адаптивно) имунно разрушаване чрез характерни микробиоми при отделни пациенти може да бъде свързано не само с прогнозата, но и с отговора или резистентността към имунотерапии с инхибитори на имунни контролни точки и други терапевтични модалности (Една проява може да бъде създаването на имунни микросреди, стимулиращи или антагонизиращи тумора, които впоследствие възникват Защитават или улесняват развитието на тумора и злокачествената прогресия.
Съответно, модулирането на преплетените параметри на (i) индуцирането на (вродено) стимулиращо тумора възпаление и (ii) бягството от (адаптивно) имунно разрушаване чрез характерни микробиоми при отделни пациенти може да бъде свързано не само с прогнозата, но и с отговора или резистентността към имунотерапии с инхибитори на имунни контролни точки и други терапевтични модалности (Една проява може да бъде създаването на имунни микросреди, стимулиращи или антагонизиращи тумора, които впоследствие се появяват, защитават или улесняват развитието на тумора и злокачествената прогресия).
Съответно, модулирането на преплетените параметри на (i) индукция на (вродено) стимулиращо тумора възпаление и (ii) бягство от (адаптивно) имунно разрушаване чрез отличителни микробиоми при отделни пациенти може да бъде свързано не само с прогноза, но и с отговор или резистентност към имунотерапии с инхибитори на имунни контролни точки и други терапевтични модалности (89, 94–96). Предварителното доказателство за концепцията идва от скорошни проучвания, показващи възстановена ефикасност на имунотерапията след трансплантации на фекална микробиота от отговорили на терапията в пациенти с меланом, който е прогресирал по време на предишно лечение с блокада на имунната контролна точка (97, 98).
Молекулярните механизми, чрез които различни и променливи компоненти на микробиома на червата системно модулират активността на адаптивната имунна система, остават постоянна мистерия, или чрез засилване на антитуморните имунни отговори, предизвикани от блокада на имунната контролна точка, или по-скоро чрез индуциране на системна или локална (интратуморна) имуносупресия. Едно скорошно проучване хвърли светлина: някои щамове на Enterococcus (и други бактерии) експресират пептидогликанова хидролиза, наречена SagA, която освобождава мукопептиди от бактериалната стена, които след това могат да циркулират системно и да активират рецептора на модела NOD2, което от своя страна повишава отговорите на Т-клетките и ефективността на имунотерапията на контролните точки (99).
Други имунорегулаторни молекули, произведени от специфични бактериални подвидове, са идентифицирани и функционално оценени, включително произведен от бактерии инозин, метаболит, ограничаващ скоростта на активността на Т клетките (100). Тези и други примери започват да очертават молекулярните механизми, чрез които полиморфните микробиоми индиректно и системно модулират туморната имунобиология, над и отвъд имунните отговори, които следват директните физически взаимодействия на бактериите с имунната система (101, 102).
Освен причинно-следствените връзки с рака на дебелото черво и меланома, демонстрираната способност на микробиома на червата да предизвиква експресията на имуномодулиращи хемокини и цитокини, които навлизат в системното кръвообращение, също очевидно е в състояние да повлияе на патогенезата на рака и отговора на терапии в други органи на тялото (94, 95).
Ясен пример се отнася до развитието на холангиокарциноми в черния дроб: чревната дисбиоза позволява навлизане и транспортиране на бактерии и бактериални продукти през порталната вена до черния дроб, където TLR4, експресиран върху хепатоцити, се задейства, за да индуцира експресия на хемокина CXCL1, който набира CXCR2-експресиращи гранулоцитни миелоидни клетки (gMDSC), които служат за потискане естествени клетки убийци, за да избегнат имунното разрушаване (103) и вероятно да предадат други отличителни способности (85). Като такъв, микробиомът на червата е ясно замесен като благоприятна характеристика, която алтернативно може да улесни или предпази от множество видове рак.
Отвъд червата: Включване на различни микробиоми в други бариерни тъкани
Почти всички тъкани и органи, които са пряко или косвено изложени на външна среда, също са хранилища на коменсални микроорганизми (104). За разлика от червата, където симбиотичната роля на микробиома в метаболизма е добре позната, нормалните и патогенни роли на резидентната микробиота в тези различни места все още се появяват.
Има очевидни специфични за органите/тъканите разлики в структурата на свързаните микробиоми в хомеостазата, стареенето и рака, както с припокриващи се, така и с отличителни видове и честоти спрямо тези на дебелото черво (104, 105). Освен това, проучванията на асоциациите предоставят все повече доказателства, че местните тумор-антагонизиращи/защитни срещу тумор-стимулиращи тъканни микробиоми, подобно на чревния микробиом, могат да модулират чувствителността и патогенезата към човешки ракови заболявания, възникващи в свързаните с тях органи (106 – 109).
Влияние на интратуморната микробиота?
И накрая, патолозите отдавна признават, че бактериите могат да бъдат открити в солидни тумори, наблюдение, което сега е подкрепено от сложни технологии за профилиране. Например, в проучване на 1526 тумора, обхващащи седем вида човешки рак (кости, мозък, гърди, бял дроб, меланом, яйчници и панкреас), всеки тип се характеризира с отличителен микробиом, разположен предимно в ракови клетки и имунни клетки. В рамките на всеки тип тумор са демонстрирани вариации в туморния микробиом и е заключено, че са свързани с клинико-патологични характеристики (110).
Микробиотата е открита по подобен начин в de novo генетично модифицирани миши модели на рак на белите дробове и панкреаса и може да се докаже, че тяхното отсъствие при свободни от микроби мишки и/или отмяната им с антибиотици нарушава туморогенезата, функционално замесвайки туморния микробиом като предшественик на стимулиращо тумора възпаление и злокачествена прогресия (111, 112).
Проучванията на асоцииране при човешки панкреатичен дуктален аденокарцином и функционални анализи чрез фекална трансплантация в мишки, носещи тумор, показват, че вариациите в туморния микробиом – и свързания с него чревен микробиом – модулират фенотиповете и оцеляването на имунната система (113). Важно предизвикателство за бъдещето ще бъде да се разширят тези последици към други видове тумори и да се разграничат потенциално отделимите приноси на конституцията и вариациите в микробиома на тумора от тези на микробиома на червата (и местната тъкан на произход), може би чрез идентифициране на специфични микробни видове, които са функционално влиятелни на едно или друго място.
Резюме
Интригуващи въпроси за бъдещето включват дали микробиотата, пребиваваща в различни тъкани или населяваща зараждащи се неоплазми, има способността да допринесе или да наруши придобиването на други отличителни способности извън имуномодулацията и геномната мутация, като по този начин влияе върху развитието и прогресията на тумора. Има доказателства, че определени бактериални видове могат директно да стимулират отличителния белег на пролиферативното сигнализиране, например в епитела на дебелото черво (88), и могат да модулират потискането на растежа чрез промяна на туморната супресорна активност в различни отделения на червата (114), докато преките ефекти върху други характерни способности, като избягване на клетъчна смърт, задействане на ангиогенеза и стимулиране на инвазия и метастази, остават неясно, както и възможността за обобщаване на тези наблюдения към множество форми на човешки рак.
Независимо от това, има все по-убедителни аргументи, че полиморфните вариации в микробиомите на червата и други органи представляват отличителна активираща характеристика за придобиване на отличителни умения (фиг. 4), дори когато се припокриват и допълват тези на геномната нестабилност и мутация и възпаление, стимулиращо тумора.
Стареещи клетки
Клетъчното стареене е типично необратима форма на спиране на пролиферацията, която вероятно се е развила като защитен механизъм за поддържане на тъканната хомеостаза, привидно като допълнителен механизъм към програмираната клетъчна смърт, която служи за инактивиране и своевременно премахване на болни, дисфункционални или по друг начин ненужни клетки. В допълнение към спирането на цикъла на клетъчно делене, програмата за стареене предизвиква промени в клетъчната морфология и метаболизъм и, най-дълбоко, активирането на секреторен фенотип, свързан със стареенето (SASP), който включва освобождаването на множество биоактивни протеини, включително хемокини.
Цитокини и протеази, чиято идентичност зависи от типа клетка и тъкан, от които възниква старееща клетка (115-117). Стареенето може да бъде предизвикано в клетките от различни условия, включително напрежения в микросредата като гладуване на хранителни вещества и увреждане на ДНК, както и увреждане на органели и клетъчна инфраструктура и дисбаланси в клетъчните сигнални мрежи (115, 117), всички от които са възникнали в контекста на наблюдаваното увеличение на честотата на стареещите клетки в различни органи по време на стареене (118, 119).
Клетъчното стареене отдавна се счита за защитен механизъм срещу неоплазия, карайки раковите клетки да претърпят стареене (120). Повечето от гореспоменатите инициатори на програмата за стареене са свързани със злокачествено заболяване, особено увреждане на ДНК в резултат на аберантна хиперпролиферация, така нареченото онкогенно индуцирано стареене поради хиперактивирано сигнализиране и индуцирано от терапия стареене в резултат на клетъчно и геномно увреждане, причинено от химиотерапия и лъчетерапия.
Наистина, има добре установени примери за защитните ползи от стареенето при ограничаване на злокачествената прогресия (118, 119). Напротив, все повече доказателства показват точно обратното: в определени контексти стареещите клетки диференцирано стимулират развитието на тумора и злокачествената прогресия (119, 121).
В едно проницателно казус, стареещите клетки в стареещи мишки са били фармакологично премахнати, по-специално изчерпвайки стареещите клетки, които характерно експресират инхибитора на клетъчния цикъл p16 – INK4a: в допълнение към забавянето на няколко свързани с възрастта симптоми, това е довело до изчерпване на стареещите клетки в стареещи мишки с намалена честота на спонтанна туморогенеза и смърт, свързана с рака (122).
Смята се, че основният механизъм, чрез който стареещите клетки насърчават туморните фенотипове, е SASP, за който е доказано, че може да медиира сигнални молекули (и протеази, които активират и/или дезактивират) по паракринен начин, за да медиира типичните способности. По този начин, в различни експериментални системи е показано, че стареещите ракови клетки допринасят по различни начини за пролиферативно сигнализиране, избягват апоптоза, индуцират ангиогенеза, стимулират инвазия и метастази и потискат туморния имунитет (116, 118, 120, 121).
Още един аспект на ефектите на стареещите ракови клетки върху раковите фенотипове включва преходни, обратими стареещи клетъчни състояния, при които стареещите ракови клетки могат да избягат от своето SASP-експресиращо, непролиферативно състояние и да възобновят клетъчната пролиферация и проявата на свързаните способности на напълно жизнеспособни онкогенни клетки (44).
Такова преходно стареене е най-добре документирано в случаи на терапевтична резистентност (44), което представлява форма на покой, който избягва терапевтичното насочване към пролифериращи ракови клетки, но може да се окаже по-широко ефективно в други етапи на развитие на тумора, злокачествена прогресия и метастази.
Освен това, способността за насърчаване на отличителен белег на стареещите клетки не се ограничава до стареещите ракови клетки. Доказано е, че асоциираните с рак фибробласти (CAF) стареят в тумори, пораждайки стареещи CAF, за които е доказано, че стимулират тумора чрез придаване на характерни способности на раковите клетки в TME (115, 116, 121).
Освен това стареещите фибробласти в нормалните тъкани, образувани отчасти от естествено стареене или екологични инсулти, участват по подобен начин в ремоделирането на тъканната микросреда чрез техния SASP, за да осигурят паракринна подкрепа за локална инвазия (така наречените „ефекти на полето“) и далечни метастази (116) на неоплазми, развиващи се наблизо.
Освен това е доказано, че стареещите фибробласти в стареещата кожа набират — чрез техния SASP — вродени имунни клетки, които са както имуносупресивни на адаптивните антитуморни имунни отговори, закрепени от CD8 Т клетки, така и стимулират растежа на кожен тумор (123), като последният ефект вероятно отразява паракринния принос на такива вродени имунни клетки (миелоидни клетки, неутрофили и макрофаги) към други характерни способности отразява.
Макар и по-малко установено, изглежда вероятно други изобилни стромални клетки, населяващи специфични туморни микросреди, да претърпят стареене, като по този начин модулират характеристиките на рака и произтичащите от това туморни фенотипове. Например, индуцираните от терапия стареещи туморни ендотелни клетки могат да засилят пролиферацията, инвазията и метастазите в модели на рак на гърдата (124, 125).
Разбира се, такива доказателства изискват изследване при други видове тумори, за да се оцени общото стареене на фибробласти, ендотелни клетки и други стромални клетки като движеща сила в развитието на тумора. Също така понастоящем неясни са регулаторните механизми и функционалните детерминанти, чрез които конкретен тип стареещи клетки в конкретен TME предизвиква тумор-стимулиращ срещу тумор-антагонизиращ SASP, който очевидно може да бъде индуциран алтернативно в същия стареещ клетъчен тип, може би от различни инициатори, когато са потопени в характерни физиологични и неопластични микросреди.
Резюме
Концепцията, че туморите се състоят от генетично трансформирани ракови клетки, които взаимодействат и се възползват от набрани и епигенетично/фенотипно повредени допълнителни (стромални) клетки, е установена като решаваща за патогенезата на рака. Съображенията, обсъдени по-горе и описани в рецензиите и докладите, цитирани тук (и другаде), доказват убедително, че стареещите клетки (независимо от клетъчния произход) трябва да бъдат разгледани за включване в списъка на функционално значимите клетки в туморната микросреда (Фиг. 5). Следователно стареещите клетки трябва да бъдат взети под внимание в търсенето на задълбочени познания за механизмите на рака. Освен това, признаването на тяхното значение мотивира вторичната цел за терапевтично насочване към стимулиращи тумора стареещи клетки от всякаква конституция, било то чрез фармакологична или имунологична аблация или чрез препрограмиране на SASP в тумор-антагонизиращи варианти (115, 121, 126).
Фигура 5

Хетерогенни подтипове ракови клетки и типове и подтипове на стромални клетки са функционално интегрирани в проявите на тумори като незаконни органи. Все повече доказателства предполагат, че стареещите клетъчни производни на много от тези клетъчни компоненти на TME и техните променливи SASPs участват в модулирането на отличителните способности и произтичащите от това туморни фенотипове. Търговските марки на графиката на рака са възприети от Hanahan и Weinberg (2).
Заключителни бележки
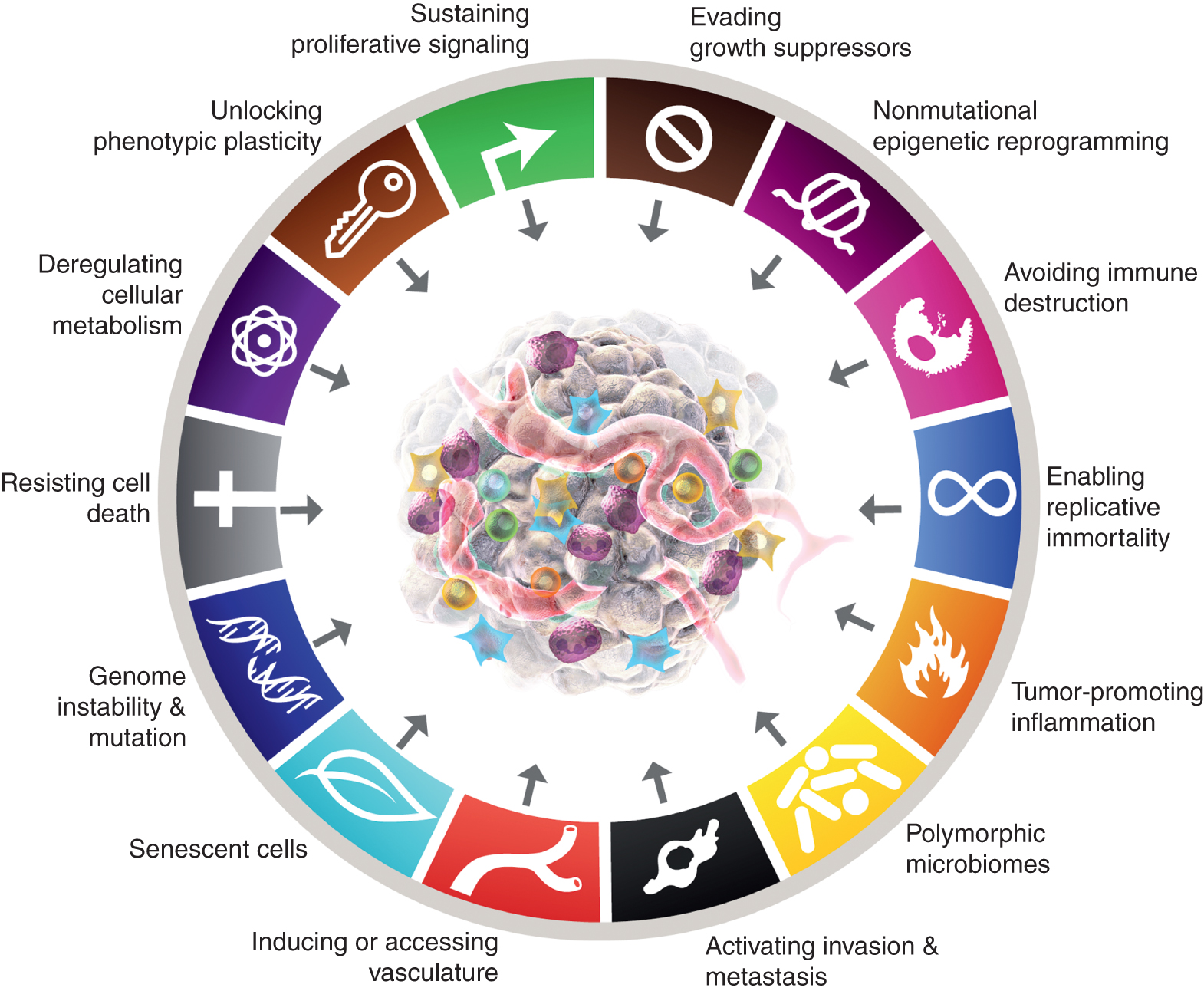
Докато осемте отличителни белега на рака и техните две поддържащи характеристики са доказали, че имат трайна евристична стойност в концептуализацията на рака, съображенията, представени по-горе, предполагат, че може да има нови аспекти на известна общност и следователно значение за по-пълното разбиране на сложността, механизмите и проявите на болестта. Чрез прилагане на показателя за забележима, ако не и пълна независимост от 10-те основни атрибута, може да се спори, че тези четири параметъра - при по-нататъшно валидиране и обобщаване извън представените казуси - могат да бъдат интегрирани в отличителните белези на схемата на рака (фиг. 6).
Следователно клетъчната пластичност може да бъде добавена към списъка с изтъкнати способности. Докато осмото ядро и тази нова способност са концептуално разграничими чрез тяхната дефиниция като отличителни белези, аспектите на тяхното регулиране са поне частично свързани в някои и може би много видове рак. Например, множество отличителни белези са координирано модулирани от канонични онкогенни драйвери в някои видове тумори, включително
- (I) KRAS ( https://cancer.sanger.ac.uk/cosmic/census-page/KRAS ),
- (II) MYC ( https://cancer.sanger.ac.uk/cosmic/census-page/MYC ),
- (III) NOTCH ( https://cancer.sanger.ac.uk/cosmic/census-page/NOTCH1 ; Ref. 127) und
- (IV) TP53 ( https://cancer.sanger.ac.uk/cosmic/census-page/TP53 )
Фигура 6
Показани са каноничните и очаквани нови допълнения към „Отличителните белези на рака“. Този документ повдига възможността, с цел стимулиране на дебат, дискусия и експериментална разработка, че някои или всички от четирите нови параметъра ще бъдат признати като родови за множество форми на човешки рак и следователно подходящи за интегриране в основната концептуализация на отличителните белези на рака. Търговските марки на графиката на рака са възприети от Hanahan и Weinberg (2).
В допълнение към добавянето на клетъчна пластичност към списъка, немутационното епигенетично препрограмиране и полиморфните вариации могат да бъдат интегрирани в органни/тъканни микробиоми като механични детерминанти – позволяващи свойства – чрез които се придобиват отличителни способности, заедно с стимулиращо тумора възпаление (самото то частично свързано с микробиома), отвъд мутациите и други аберации, които проявяват споменатите онкогенни драйвери по-горе.
И накрая, стареещи клетки от различен произход - включително ракови клетки и различни стромални клетки - които функционално допринасят за развитието и злокачествената прогресия на рака, макар и по значително различни начини от тези на техните нестареещи събратя, могат да бъдат включени като общи компоненти на TME. В обобщение, предвижда се, че разполагането на тези предварителни „експериментални балони“ ще стимулира дебат, дискусия и по-нататъшно експериментално изследване в общността за изследване на рака относно определящите концептуални параметри на биологията, генетиката и патогенезата на рака.
Референции
- Hanahan D , Weinberg RA . The hallmarks of cancer. Cell 2000;100:57–70.
- Hanahan D , Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Yuan S , Norgard RJ , Stanger BZ . Cellular plasticity in cancer. Cancer Discov 2019;9:837–51.
- Barker N , Ridgway RA , van Es JH , van de Wetering M , Begthel H , van den Born M et al . Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608–11.
- Perekatt AO , Shah PP , Cheung S , Jariwala N , Wu A , Gandhi V et al . SMAD4 suppresses WNT-driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res 2018;78:4878–90.
- Shih IM , Wang TL , Traverso G , Romans K , Hamilton SR , Ben-Sasson S et al . Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A 2001;98:2640–5.
- Ordóñez-Morán P , Dafflon C , Imajo M , Nishida E , Huelsken J . HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 2015;28:815–29.
- Tan SH , Barker N . Stemming colorectal cancer growth and metastasis: HOXA5 forces cancer stem cells to differentiate. Cancer Cell 2015;28:683–5.
- Köhler C , Nittner D , Rambow F , Radaelli E , Stanchi F , Vandamme N et al . Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 2017;21:679–93.
- Shah M , Bhoumik A , Goel V , Dewing A , Breitwieser W , Kluger H et al . A role for ATF2 in regulating MITF and melanoma development. PLoS Genet 2010;6:e1001258.
- Claps G , Cheli Y , Zhang T , Scortegagna M , Lau E , Kim H et al . A transcriptionally inactive ATF2 variant drives melanomagenesis. Cell Rep 2016;15:1884–92.
- Saghafinia S , Homicsko K , Di Domenico A , Wullschleger S , Perren A , Marinoni I et al . Cancer cells retrace a stepwise differentiation program during malignant progression. Cancer Discov 2021;11:2638–57.
- Yu X-X , Qiu W-L , Yang L , Zhang Y , He M-Y , Li L-C et al . Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J 2019;38:e100164.
- de Thé H . Differentiation therapy revisited. Nat Rev Cancer 2018;18:117–27.
- He LZ , Merghoub T , Pandolfi PP . In vivo analysis of the molecular pathogenesis of acute promyelocytic leukemia in the mouse and its therapeutic implications. Oncogene 1999;18:5278–92.
- Warrell RP , de Thé H , Wang ZY , Degos L . Acute promyelocytic leukemia. N Engl J Med 1993;329:177–89.
- Bots M , Verbrugge I , Martin BP , Salmon JM , Ghisi M , Baker A et al . Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014;123:1341–52.
- Ferrara FF , Fazi F , Bianchini A , Padula F , Gelmetti V , Minucci S et al . Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res 2001;61:2–7.
- Kaufman CK , Mosimann C , Fan ZP , Yang S , Thomas AJ , Ablain J et al . A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016;351:aad2197.
- Morris JP , Yashinskie JJ , Koche R , Chandwani R , Tian S , Chen C-C et al . α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019;573:595–9.
- Saha SK , Parachoniak CA , Ghanta KS , Fitamant J , Ross KN , Najem MS et al . Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110–4.
- Dang L , Su S-SM . Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem 2017;86:305–31.
- Waitkus MS , Diplas BH , Yan H . Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018;34:186–95.
- Phan TG , Croucher PI . The dormant cancer cell life cycle. Nat Rev Cancer 2020;20:398–411.
- Jiang M , Azevedo-Pouly AC , Deering TG , Hoang CQ , DiRenzo D , Hess DA et al . MIST1 and PTF1 collaborate in feed-forward regulatory loops that maintain the pancreatic acinar phenotype in adult mice. Mol Cell Biol 2016;36:2945–55.
- Krah NM , Narayanan SM , Yugawa DE , Straley JA , Wright CVE , MacDonald RJ et al . Prevention and reversion of pancreatic tumorigenesis through a differentiation-based mechanism. Dev Cell 2019;50:744–54.
- Krah NM , De La O J-P , Swift GH , Hoang CQ , Willet SG , Chen Pan F et al . The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015;4:e07125.
- Shi G , DiRenzo D , Qu C , Barney D , Miley D , Konieczny SF . Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 2013;32:1950–8.
- Kopp JL , von Figura G , Mayes E , Liu F-F , Dubois CL , Morris JP et al . Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50.
- Julian LM , McDonald AC , Stanford WL . Direct reprogramming with SOX factors: masters of cell fate. Curr Opin Genet Dev 2017;46:24–36.
- Grimm D , Bauer J , Wise P , Krüger M , Simonsen U , Wehland M et al . The role of SOX family members in solid tumours and metastasis. Semin Cancer Biol 2020;67:122–53.
- Mu P , Zhang Z , Benelli M , Karthaus WR , Hoover E , Chen C-C et al . SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84–8.
- Von Hoff DD , LoRusso PM , Rudin CM , Reddy JC , Yauch RL , Tibes R et al . Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72.
- Biehs B , Dijkgraaf GJP , Piskol R , Alicke B , Boumahdi S , Peale F et al . A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018;562:429–33.
- Boumahdi S , de Sauvage FJ . The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19:39–56.
- Groves SM , Ireland A , Liu Q , Simmons AJ , Lau K , Iams WT et al . Cancer Hallmarks Define a Continuum of Plastic Cell States between Small Cell Lung Cancer Archetypes [Internet]. Systems Biology; 2021 Jan. Available from: http://biorxiv.org/lookup/doi/10.1101/2021.01.22.427865.
- LaFave LM , Kartha VK , Ma S , Meli K , Del Priore I , Lareau C et al . Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 2020;38:212–28.
- Marjanovic ND , Hofree M , Chan JE , Canner D , Wu K , Trakala M et al . Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 2020;38:229–46.
- Drapkin BJ , Minna JD . Studying lineage plasticity one cell at a time. Cancer Cell 2020;38:150–2.
- Inoue Y , Nikolic A , Farnsworth D , Liu A , Ladanyi M , Somwar R et al . Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer [Internet]. Cancer Biology; 2020 Nov. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.11.12.368522.
- Dravis C , Chung C-Y , Lytle NK , Herrera-Valdez J , Luna G , Trejo CL et al . Epigenetic and transcriptomic profiling of mammary gland development and tumor models disclose regulators of cell state plasticity. Cancer Cell 2018;34:466–82.
- Malta TM , Sokolov A , Gentles AJ , Burzykowski T , Poisson L , Weinstein JN et al . Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–54.
- Miao Z-F , Lewis MA , Cho CJ , Adkins-Threats M , Park D , Brown JW et al . A dedicated evolutionarily conserved molecular network licenses differentiated cells to return to the cell cycle. Dev Cell 2020;55:178–94.
- De Blander H , Morel A-P , Senaratne AP , Ouzounova M , Puisieux A . Cellular plasticity: a route to senescence exit and tumorigenesis. Cancers 2021;13:4561.
- Merrell AJ , Stanger BZ . Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 2016;17:413–25.
- Baylin SB , Jones PA . Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 2016;8:a019505.
- Flavahan WA , Gaskell E , Bernstein BE . Epigenetic plasticity and the hallmarks of cancer. Science 2017;357:eaal2380.
- Jones PA , Issa J-PJ , Baylin S . Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630–41.
- Huang S . Tumor progression: Chance and necessity in Darwinian and Lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol 2012;110:69–86.
- Darwiche N . Epigenetic mechanisms and the hallmarks of cancer: an intimate affair. Am J Cancer Res 2020;10:1954–78.
- Feng Y , Liu X , Pauklin S . 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell 2021;12:440–54.
- Nam AS , Chaligne R , Landau DA . Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet 2021;22:3–18.
- Bitman-Lotan E , Orian A . Nuclear organization and regulation of the differentiated state. Cell Mol Life Sci CMLS 2021;78:3141–58.
- Goldberg AD , Allis CD , Bernstein E . Epigenetics: a landscape takes shape. Cell 2007;128:635–8.
- Zeng Y , Chen T . DNA methylation reprogramming during mammalian development. Genes 2019;10:257.
- Hegde AN , Smith SG . Recent developments in transcriptional and translational regulation underlying long-term synaptic plasticity and memory. Learn Mem 2019;26:307–17.
- Kim S , Kaang B-K . Epigenetic regulation and chromatin remodeling in learning and memory. Exp Mol Med 2017;49:e281.
- Thienpont B , Van Dyck L , Lambrechts D . Tumors smother their epigenome. Mol Cell Oncol 2016;3:e1240549.
- Gameiro PA , Struhl K . Nutrient deprivation elicits a transcriptional and translational inflammatory response coupled to decreased protein synthesis. Cell Rep 2018;24:1415–24.
- Lin GL , Monje M . Understanding the deadly silence of posterior fossa A ependymoma. Mol Cell 2020;78:999–1001.
- Michealraj KA , Kumar SA , Kim LJY , Cavalli FMG , Przelicki D , Wojcik JB et al . Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell 2020;181:1329–45.
- Bakir B , Chiarella AM , Pitarresi JR , Rustgi AK . EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol 2020;30:764–76.
- Gupta PB , Pastushenko I , Skibinski A , Blanpain C , Kuperwasser C . Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 2019;24:65–78.
- Lambert AW , Weinberg RA . Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer 2021;21:325–38.
- Lindner P , Paul S , Eckstein M , Hampel C , Muenzner JK , Erlenbach-Wuensch K et al . EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis 2020;11:147.
- Javaid S , Zhang J , Anderssen E , Black JC , Wittner BS , Tajima K et al . Dynamic chromatin modification sustains epithelial-mesenchymal transition following inducible expression of Snail-1. Cell Rep 2013;5:1679–89.
- Serrano-Gomez SJ , Maziveyi M , Alahari SK . Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 2016;15:18.
- Skrypek N , Goossens S , De Smedt E , Vandamme N , Berx G . Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet TIG 2017;33:943–59.
- Li L , Hanahan D . Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 2013;153:86–100.
- Li L , Zeng Q , Bhutkar A , Galván JA , Karamitopoulou E , Noordermeer D et al . GKAP acts as a genetic modulator of NMDAR signaling to govern invasive tumor growth. Cancer Cell 2018;33:736–51.
- Mohammadi H , Sahai E . Mechanisms and impact of altered tumour mechanics. Nat Cell Biol 2018;20:766–74.
- Odenthal J , Takes R , Friedl P . Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis 2016;37:1117–28.
- Torres CM , Biran A , Burney MJ , Patel H , Henser-Brownhill T , Cohen A-HS et al . The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 2016;353:aaf1644.
- Puram SV , Tirosh I , Parikh AS , Patel AP , Yizhak K , Gillespie S et al . Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171:1611–24.
- Kinker GS , Greenwald AC , Tal R , Orlova Z , Cuoco MS , McFarland JM et al . Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 2020;52:1208–18.
- Murtha M , Esteller M . Extraordinary cancer epigenomics: thinking outside the classical coding and promoter box. Trends Cancer 2016;2:572–84.
- Nebbioso A , Tambaro FP , Dell’Aversana C , Altucci L . Cancer epigenetics: moving forward. PLoS Genet 2018;14:e1007362.
- Tavernari D , Battistello E , Dheilly E , Petruzzella AS , Mina M , Sordet-Dessimoz J et al . Non-genetic evolution drives lung adenocarcinoma spatial heterogeneity and progression. Cancer Discov 2021;11:1490–507.
- Heyn H , Vidal E , Ferreira HJ , Vizoso M , Sayols S , Gomez A et al . Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 2016;17:11.
- Saghafinia S , Mina M , Riggi N , Hanahan D , Ciriello G . Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 2018;25:1066–80.
- Audia JE , Campbell RM . Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8:a019521.
- Corces MR , Granja JM , Shams S , Louie BH , Seoane JA , Zhou W et al . The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898.
- Esteve-Puig R , Bueno-Costa A , Esteller M . Writers, readers and erasers of RNA modifications in cancer. Cancer Lett 2020;474:127–37.
- Janin M , Coll-SanMartin L , Esteller M . Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer 2020;19:70.
- Hanahan D , Coussens LM . Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–22.
- Lu Z , Zou J , Li S , Topper MJ , Tao Y , Zhang H et al . Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020;579:284–90.
- Thomas S , Izard J , Walsh E , Batich K , Chongsathidkiet P , Clarke G et al . The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res 2017;77:1783–812.
- Dzutsev A , Badger JH , Perez-Chanona E , Roy S , Salcedo R , Smith CK et al . Microbes and cancer. Annu Rev Immunol 2017;35:199–228.
- Helmink BA , Khan MAW , Hermann A , Gopalakrishnan V , Wargo JA . The microbiome, cancer, and cancer therapy. Nat Med 2019;25:377–88.
- Sears CL , Garrett WS . Microbes, microbiota, and colon cancer. Cell Host Microbe 2014;15:317–28.
- Pleguezuelos-Manzano C , Puschhof J , Rosendahl Huber A , van Hoeck A , Wood HM , Nomburg J et al . Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020;580:269–73.
- Okumura S , Konishi Y , Narukawa M , Sugiura Y , Yoshimoto S , Arai Y et al . Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat Commun 2021;12:5674.
- Salvi PS , Cowles RA . Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in homeostasis and disease. Cells 2021;10:1775.
- Fessler J , Matson V , Gajewski TF . Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 2019;7:108.
- Gopalakrishnan V , Helmink BA , Spencer CN , Reuben A , Wargo JA . The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 2018;33:570–80.
- Zitvogel L , Ma Y , Raoult D , Kroemer G , Gajewski TF . The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–70.
- Baruch EN , Youngster I , Ben-Betzalel G , Ortenberg R , Lahat A , Katz L et al . Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021;371:602–9.
- Davar D , Dzutsev AK , McCulloch JA , Rodrigues RR , Chauvin J-M , Morrison RM et al . Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021;371:595–602.
- Griffin ME , Espinosa J , Becker JL , Luo J-D , Carroll TS , Jha JK et al . Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 2021;373:1040–6.
- Mager LF , Burkhard R , Pett N , Cooke NCA , Brown K , Ramay H et al . Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369:1481–9.
- Ansaldo E , Belkaid Y . How microbiota improve immunotherapy. Science 2021;373:966–7.
- Ansaldo E , Farley TK , Belkaid Y . Control of immunity by the microbiota. Annu Rev Immunol 2021;39:449–79.
- Zhang Q , Ma C , Duan Y , Heinrich B , Rosato U , Diggs LP et al . Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov 2021;11:1248–67.
- Ding T , Schloss PD . Dynamics and associations of microbial community types across the human body. Nature 2014;509:357–60.
- Byrd AL , Liu M , Fujimura KE , Lyalina S , Nagarkar DR , Charbit B et al . Gut microbiome stability and dynamics in healthy donors and patients with non-gastrointestinal cancers. J Exp Med 2021;218:e20200606.
- Healy CM , Moran GP . The microbiome and oral cancer: more questions than answers. Oral Oncol 2019;89:30-3.
- Swaney MH , Kalan LR . Living in your skin: microbes, molecules and mechanisms. Infect Immun 2021;89:e00695–20.
- Willis JR , Gabaldón T . The human oral microbiome in health and disease: from sequences to ecosystems. Microorganisms 2020;8:308.
- Xu J , Peng J-J , Yang W , Fu K , Zhang Y . Vaginal microbiomes and ovarian cancer: a review. Am J Cancer Res 2020;10:743–56.
- Nejman D , Livyatan I , Fuks G , Gavert N , Zwang Y , Geller LT et al . The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80.
- Jin C , Lagoudas GK , Zhao C , Bullman S , Bhutkar A , Hu B et al . Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019;176:998–1013.
- Pushalkar S , Hundeyin M , Daley D , Zambirinis CP , Kurz E , Mishra A et al . The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 2018;8:403–16.
- McAllister F , Khan MAW , Helmink B , Wargo JA . The tumor microbiome in pancreatic cancer: bacteria and beyond. Cancer Cell 2019;36:577–9.
- Kadosh E , Snir-Alkalay I , Venkatachalam A , May S , Lasry A , Elyada E et al . The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020;586:133–8.
- Birch J , Gil J . Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34:1565–76.
- Faget DV , Ren Q , Stewart SA . Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 2019;19:439–53.
- Gorgoulis V , Adams PD , Alimonti A , Bennett DC , Bischof O , Bishop C et al . Cellular senescence: defining a path forward. Cell 2019;179:813–27.
- He S , Sharpless NE . Senescence in health and disease. Cell 2017;169:1000–11.
- Kowald A , Passos JF , Kirkwood TBL . On the evolution of cellular senescence. Aging Cell 2020;19:e13270.
- Lee S , Schmitt CA . The dynamic nature of senescence in cancer. Nat Cell Biol 2019;21:94–101.
- Wang B , Kohli J , Demaria M . Senescent cells in cancer therapy: friends or foes? Trends Cancer 2020;6:838–57.
- Baker DJ , Childs BG , Durik M , Wijers ME , Sieben CJ , Zhong J et al . Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–9.
- Ruhland MK , Loza AJ , Capietto A-H , Luo X , Knolhoff BL , Flanagan KC et al . Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 2016;7:11762.
- Hwang HJ , Lee Y-R , Kang D , Lee HC , Seo HR , Ryu J-K et al . Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett 2020;490:100–10.
- Wang D , Xiao F , Feng Z , Li M , Kong L , Huang L et al . Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res 2020;22:103.
- Amor C , Feucht J , Leibold J , Ho Y-J , Zhu C , Alonso-Curbelo D et al . Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020;583:127–32.
- Aster JC , Pear WS , Blacklow SC . The varied roles of notch in cancer. Annu Rev Pathol 2017;12:245–75.
- Kuczynski EA , Vermeulen PB , Pezzella F , Kerbel RS , Reynolds AR . Vessel co-option in cancer. Nat Rev Clin Oncol 2019;16:469–93.
Google ScholarCrossref