 Suche
Suche
 Mein Konto
Mein Konto
Charakteristika Raka: Nové dimenze

Předmluva Charakteristiky konceptualizace rakoviny jsou heuristickým nástrojem pro destilaci obrovské složitosti fenotypů a genotypů rakoviny do předběžného souboru základních principů. Jak znalosti mechanismů rakoviny pokročily, objevily se další aspekty onemocnění jako potenciální vylepšení. To vyvolává vyhlídku, že fenotypová plasticita a neuspořádaná diferenciace jsou výraznou charakteristickou schopností a že nemutační epigenetické přeprogramování a polymorfní mikrobiomy představují charakteristické umožňující vlastnosti, které usnadňují získání charakteristických schopností. Dále mohou být senescentní buňky různého původu přidány do seznamu funkčně důležitých typů buněk v mikroprostředí nádoru. To znamená, že rakovina je děsivá v...

Charakteristika Raka: Nové dimenze
Předmluva
Charakteristiky konceptualizace rakoviny jsou heuristickým nástrojem pro destilaci obrovské složitosti rakovinných fenotypů a genotypů do předběžného souboru základních principů. Jak znalosti mechanismů rakoviny pokročily, objevily se další aspekty onemocnění jako potenciální vylepšení. To vyvolává vyhlídku, že fenotypová plasticita a neuspořádaná diferenciace jsou výraznou charakteristickou schopností a že nemutační epigenetické přeprogramování a polymorfní mikrobiomy představují charakteristické umožňující vlastnosti, které usnadňují získání charakteristických schopností. Dále mohou být senescentní buňky různého původu přidány do seznamu funkčně důležitých typů buněk v mikroprostředí nádoru.
Význam
Rakovina je děsivá v šíři a rozsahu své rozmanitosti, která zahrnuje genetiku, buněčnou a tkáňovou biologii, patologii a reakci na terapii. Stále výkonnější experimentální a výpočetní nástroje a technologie poskytují lavinu „velkých dat“ o nesčetných projevech onemocnění, které rakovina zahrnuje. Integrační koncept ztělesněný ve znacích rakoviny pomáhá destilovat tuto složitost do stále logičtější vědy a předběžné nové dimenze prezentované v této perspektivě mohou přidat hodnotu tomuto úsilí lépe porozumět mechanismům karcinogeneze a maligní progrese a aplikovat tyto znalosti na medicínu rakoviny.
zavedení
Charakteristické znaky rakoviny byly navrženy jako soubor funkčních schopností, které lidské buňky získávají, když se pohybují od normálního stavu k neoplastickému růstu, konkrétněji schopnosti kritické pro jejich schopnost tvořit maligní nádory. V těchto článcích ( 1, 2 ), Bob Weinberg a já jsme uvedli to, co jsme si představovali jako společné rysy, které sjednocují všechny typy rakovinných buněk na úrovni buněčného fenotypu. Záměrem bylo poskytnout koncepční rámec, který by umožnil racionalizaci komplexních fenotypů různých typů a variant lidských nádorů ve vztahu ke společnému souboru základních buněčných parametrů. Původně jsme si představovali doplňkové zahrnutí šesti různých funkcí značky a později jsme tento počet rozšířili na osm.
Tato formulace byla ovlivněna zjištěním, že lidské rakoviny se vyvíjejí jako produkty vícestupňových procesů a že získání těchto funkčních schopností lze nějakým způsobem připsat odlišným krokům patogeneze nádoru. Různorodost maligní patogeneze, zahrnující více typů nádorů a stále větší množství podtypů, zahrnuje různé aberace (a tím i získané schopnosti a vlastnosti), které jsou výsledkem tkáňově specifických bariér, které jsou během určitých cest tumorigeneze nutně obcházeny. Ačkoli uznáváme, že takové specializované mechanismy mohou být užitečné, omezili jsme označení charakteristických znaků na parametry, které mají široký dopad na celé spektrum lidských rakovin.
Mezi osm charakteristických znaků v současné době patří (obr.1, vlevo) získané schopnosti udržovat proliferativní signalizaci, vyhýbat se supresorům růstu, odolávat buněčné smrti, umožnit replikační nesmrtelnost, indukovat/vstupovat do cév, aktivovat invazi a metastázy, přeprogramovat buněčný metabolismus a vyhnout se destrukci imunitního systému. V nejnovějším zpracování tohoto konceptu (2) byly deregulace buněčného metabolismu a vyhýbání se destrukci imunitního systému vymezeny jako „vznikající charakteristické znaky“, ale nyní, o jedenáct let později, je zřejmé, že podobně jako původních šest je lze považovat za základní charakteristické znaky rakoviny a jako takové jsou zahrnuty do současného příběhu (obr. 1 vlevo).
Obrázek 1
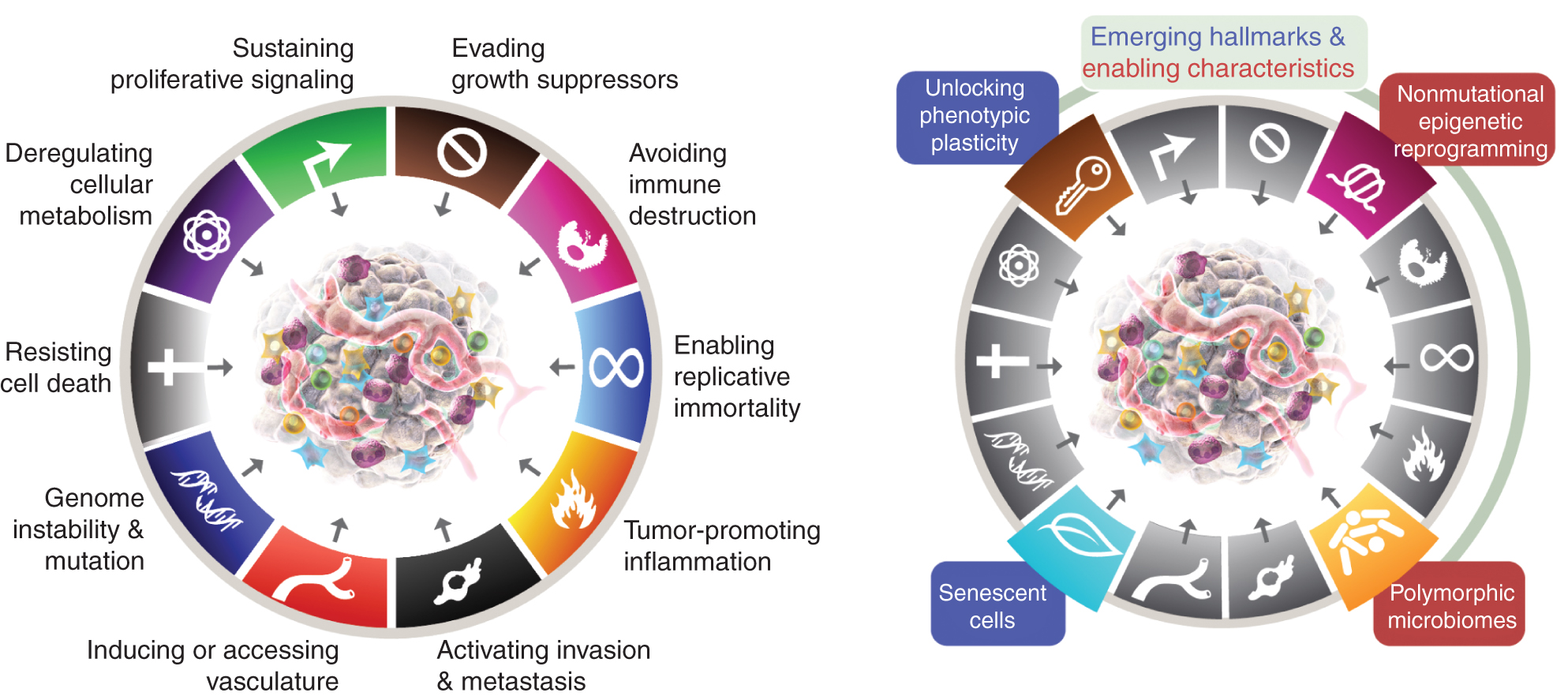
Charakteristické znaky Raka v současnosti ztělesňují osm výrazných schopností a dvě podpůrné vlastnosti. Kromě šesti získaných schopností – charakteristických znaků rakoviny – navržených v roce 2000 (1), byly dostatečně ověřeny dva předběžné „emergentní znaky“ představené v roce 2011 (2) – buněčná energetika (nyní častěji označovaná jako „přeprogramování buněčného metabolismu“) a „vyhýbání se destrukci imunitního systému“ – dostatečně ověřené, aby mohly být považovány za součást základního souboru.
Vzhledem k rostoucímu poznání, že nádory mohou být adekvátně vaskularizovány, buď zapnutím angiogeneze nebo kooptací normální tkáňové vaskulatury (128), je tento charakteristický znak také šířeji definován jako schopnost indukovat nebo jinak zpřístupnit vaskulaturu podporující růst nádoru primárně prostřednictvím invaze a metastáz.
Pokračování z roku 2011 také zahrnovalo „zánět podporující nádor“ jako druhou umožňující vlastnost, doplňující zastřešující „genomovou nestabilitu a mutaci“, které se společně zásadně podílely na aktivaci osmi charakteristických (funkčních) schopností potřebných pro růst a progresi nádoru. Je pravda, že tato recenze obsahuje další navrhované nové charakteristické znaky a funkce, včetně „odemknutí fenotypové plasticity“, „nemutačního epigenetického přeprogramování“, „polymorfních mikrobiomů“ a „stárnoucích buněk“. Ochranné známky grafiky rakoviny byly převzaty od Hanahana a Weinberga (2).
Jak jsme v té době poznamenali, tyto charakteristické rysy samy o sobě nemohou řešit složitost patogeneze rakoviny, tj. přesné molekulární a buněčné mechanismy, které umožňují vyvíjejícím se preneoplastickým buňkám vyvinout a získat tyto aberantní fenotypové schopnosti v průběhu tumorigeneze a maligní progrese.
V souladu s tím jsme do diskuse přidali další koncept prezentovaný jako „umožňující rysy“, důsledky aberantního stavu novotvaru, které poskytují prostředky, kterými mohou rakovinné buňky a nádory získat tyto funkční vlastnosti. Umožňující vlastnosti se jako takové odrážejí spíše v molekulárních a buněčných mechanismech, jejichž prostřednictvím se získávají charakteristické znaky, než ve výše uvedených osmi dovednostech samotných. Tyto dva aktivační procesy byly nestabilita genomu a zánět podporující nádor.
Dále jsme uznali, že nádorové mikroprostředí (TME), zde definované jako složené z heterogenních a interaktivních populací rakovinných buněk a rakovinných kmenových buněk spolu s různými typy rekrutovaných stromálních buněk – transformovaný parenchym a související stroma – je nyní široce uznáváno, že hraje zásadní roli v tumorigenezi a maligní progresi.
Vzhledem k trvalému zájmu o tyto formulace a našemu trvalému záměru podporovat pokračující diskusi a zpřesňování Hallmarks schématu je vhodné zvážit často kladenou otázku: Existují další rysy tohoto koncepčního modelu, které by bylo možné začlenit s přihlédnutím k nutnosti toto zajistit? že jsou široce použitelné v celém spektru lidských rakovin? V souladu s tím uvádím několik potenciálních nových charakteristických znaků a funkcí, které by mohly být časem integrovány jako základní součásti charakteristických znaků konceptualizace rakoviny.
Tyto parametry jsou „odemykání fenotypové plasticity“, „nemutační epigenetické přeprogramování“, „polymorfní mikrobiomy“ a „senescentní buňky“ (obr. 1 vpravo). Důležité je, že příklady uvedené na podporu těchto tezí jsou ilustrativní, ale v žádném případě nejsou vyčerpávající, protože existuje rostoucí a stále přesvědčivější množství publikovaných důkazů na podporu každé viněty.
Odpichová fenotypová plasticita
Během organogeneze je vývoj, determinace a organizace buněk do tkání pro provádění homeostatických funkcí doprovázena terminální diferenciací, přičemž progenitorové buňky přestávají růst, někdy ireverzibilně, jak tyto procesy kulminují. Jako takový je konečný výsledek buněčné diferenciace ve většině případů antiproliferativní, tvořící jasnou bariéru pro pokračující proliferaci nezbytnou pro neoplazii.
Přibývá důkazů, že uvolnění normálně omezené schopnosti fenotypové plasticity obejít nebo uniknout stavu terminální diferenciace je kritickou složkou patogeneze rakoviny (3). Tato plasticita může působit v několika projevech (obr. 2). Tak vznikající rakovinné buňky, které pocházejí z normální buňky, která se vyvinula cestou, která se blíží nebo nabývá plně diferencovaného stavu, mohou zvrátit průběh dediferenciací zpět na stavy podobné progenitorovým buněčným stavům.
Naopak neoplastické buňky pocházející z progenitorových buněk určených k tomu, aby následovaly dráhu vedoucí k terminální diferenciaci, mohou tento proces zkrátit a udržet expandující rakovinné buňky v částečně diferencovaném stavu podobném progenitoru. Alternativně může dojít k transdiferenciaci, kdy buňky původně zapojené do jedné diferenciační dráhy přejdou na zcela odlišný vývojový program a tím získají tkáňově specifické vlastnosti, které nebyly předem určeny jejich normálními buňkami původu.
Následující příklady podporují argument, že různé formy buněčné plasticity odhalují fenotypovou plasticitu. Vlevo je fenotypová plasticita pravděpodobně získaná charakteristická schopnost, která umožňuje různé poruchy diferenciace buněk, včetně (i) dediferenciace ze zralých do progenitorových stavů, (ii) zastavené (terminální) diferenciace od stavů progenitorových buněk a (iii) transdiferenciace do jiných buněčných linií. Vpravo jsou znázorněny tři prominentní způsoby zhoršené diferenciace, které jsou nedílnou součástí patogeneze rakoviny.
Rozdílným zvrácením normální diferenciace progenitorových buněk na zralé buňky ve vývojových liniích je usnadněna tumorigeneze a maligní progrese vznikající z buněk původu v takových drahách. Ochranné známky grafiky rakoviny byly převzaty od Hanahana a Weinberga (2).
Obrázek 2

Dediferenciace
Karcinogeneze tlustého střeva je příkladem narušené diferenciace, protože existuje teleologická potřeba, aby počínající rakovinné buňky unikly z dopravního pásu terminální diferenciace a exfoliace, k čemuž by v zásadě mohlo dojít dediferenciací epiteliálních buněk tlustého střeva, které se ještě terminálně nediferencovaly, nebo zastavenou diferenciací progenitorových/kmenových buněk v těchto odlišných kryptách, které dávají vzniknout těmto odlišným kryptám. Jak diferencované buňky, tak kmenové buňky byly implikovány jako buňky původu pro rakovinu tlustého střeva (4 – 6).
Dva vývojové transkripční faktory (TF), homeobox protein HOXA5 a SMAD4, druhý uvedený v BMP signalizaci, jsou vysoce exprimovány v diferencujících se epiteliálních buňkách tlustého střeva a jsou typicky ztraceny u pokročilých karcinomů tlustého střeva, které charakteristicky exprimují markery kmenových a progenitorových buněk. Funkční poruchy na myších modelech ukázaly, že nucená exprese HOXA5 v buňkách rakoviny tlustého střeva obnovuje diferenciační markery, potlačuje fenotypy kmenových buněk a zhoršuje invazi a metastázy, což poskytuje zdůvodnění pro její charakteristickou downregulaci (7, 8).
Na rozdíl od toho SMAD4 vynucuje jak diferenciaci, tak potlačení proliferace řízené onkogenní signalizací WNT, což je odhaleno umělou ztrátou exprese SMAD4, což poskytuje vysvětlení pro jeho ztrátu exprese umožňující dediferenciaci a následně hyperproliferaci řízenou WNT (5).
Je pozoruhodné, že ztráta těchto dvou „potlačovačů diferenciace“ s výslednou dediferenciací je spojena se získáním dalších charakteristických schopností, jakož i dalších regulátorů vyvolávajících punc, což komplikuje přísnou definici tohoto prozatímního puncu jako oddělitelného a nezávislého.
Další řada důkazů se týká suprimované exprese MITF hlavního regulátoru diferenciace melanocytů, který se zdá být zapojen do geneze agresivních forem maligního melanomu. Ztráta této vývojové TF je spojena s reaktivací progenitorových genů neurální lišty a downregulací genů, které charakterizují plně diferencované melanocyty. Znovuobjevení genů neurální lišty ukazuje, že se tyto buňky vracejí do progenitorového stavu, ze kterého vývojově vznikají melanocyty.
Kromě toho studie sledování linie melanomů indukovaných BRAF prokázala zralé pigmentované melanocyty jako buňky původu, které podléhají dediferenciaci v průběhu tumorigeneze (9). Je pozoruhodné, že mutantní onkogen BRAF, který se nachází ve více než polovině kožních melanomů, indukuje hyperproliferaci, která předchází a může být proto mechanicky oddělena od následné dediferenciace, která vzniká downregulací MITF.
Další studie funkčně implikovala upregulaci vývojového TF ATF2, jehož charakteristická exprese v myších a lidských melanomech nepřímo potlačuje MITF1, současně s maligní progresí následně dediferencovaných melanomových buněk (10). Naopak exprese mutantních forem ATF2 v melanomech, které nemohou potlačit MITF, vede k dobře diferencovaným melanomům (11).
Kromě toho nedávná studie (12) spojila dediferenciaci linií s maligní progresí novotvarů buněk pankreatických ostrůvků ke karcinomům náchylným ke vzniku metastáz; tyto neuroendokrinní buňky a odvozené nádory pocházejí z vývojové linie odlišné od linie, která generuje mnohem větší počet sousedních buněk, které tvoří exokrinní a pankreas a výsledné duktální adenokarcinomy.
Je pozoruhodné, že vícestupňová diferenciační dráha od progenitorových buněk ostrůvků ke zralým β-buňkám byla důkladně charakterizována (13). Srovnávací profilování transkriptomů ukazuje, že adenomové ostrůvkové nádory jsou nejpodobnější nezralým, ale diferencovaným β-buňkám produkujícím inzulín, zatímco invazivní karcinomy jsou nejpodobnější prekurzorům embryonálních ostrůvkových buněk. Progrese do špatně diferencovaných karcinomů zahrnuje počáteční krok dediferenciace, která zpočátku nezahrnuje zvýšenou proliferaci nebo sníženou apoptózu ve srovnání s dobře diferencovanými adenomy, přičemž oba mají tendenci se vyskytovat později.
Diskrétní krok dediferenciace tedy není řízen pozorovatelnými změnami v charakteristických rysech trvalé proliferace a odolnosti vůči apoptóze. Upregulace miRNA, která se dříve podílela na specifikaci stavu progenitoru ostrůvků, je spíše ta, která je downregulována během terminální diferenciace β-buněk, 12).
Zablokovaná diferenciace
Zatímco výše uvedené příklady ilustrují, jak může suprese exprese diferenciačního faktoru usnadnit tumorigenezi tím, že umožní lépe diferencovaným buňkám dediferenciovat se na progenitorové buňky, v jiných případech mohou neúplně diferencované progenitorové buňky trpět regulačními změnami, které aktivně blokují jejich další progresi do plně diferencovaných, typicky neproliferačních stavů.
Již dlouho bylo zdokumentováno, že akutní promyelocytární leukémie (APL) je důsledkem chromozomální translokace, která fúzuje lokus PML s genem kódujícím nukleární receptor kyseliny retinové (RARα). Myeloidní progenitorové buňky nesoucí takové translokace zjevně nejsou schopny pokračovat ve své obvyklé terminální diferenciaci na granulocyty, což vede k buňkám uvězněným v proliferativním, promyelocytům podobnému progenitorovému stadiu (14).
Důkaz koncepce pro toto schéma pochází z léčby kultivovaných buněk APL, myších modelů onemocnění a postižených pacientů kyselinou retinovou, ligandem RARα; Tato terapeutická léčba způsobuje, že se neoplastické APL buňky diferencují na zjevně zralé, neproliferující granulocyty, čímž se zkracuje jejich progresivní proliferativní expanze (14–16).
Variace na toto téma se týká jiné formy akutní myeloidní leukémie, která nese translokaci t(8;21), která produkuje fúzní protein AML1-ETO. Tento protein samotný může transformovat myeloidní progenitory, alespoň částečně blokováním jejich diferenciace. Terapeutická intervence na myších modelech a pacientech s farmakologickým inhibitorem chromatin-modifikující histondeacetylázy (HDAC) způsobuje obnovení diferenciace buněk myeloidní leukémie na buňky se zralejší myeloidní buněčnou morfologií. Doprovázející tuto reakci je snížení proliferační kapacity, a tím zhoršení progrese této leukémie (17, 18).
Třetí příklad u melanomu zahrnuje vývojový TF, SOX10, který je normálně downregulován během diferenciace melanocytů. Studie zisku a ztráty funkce na modelu zebrafish melanomů indukovaných BRAF ukázaly, že abnormálně udržovaná exprese SOX10 blokuje diferenciaci nervových progenitorových buněk na melanocyty, což umožňuje tvorbu melanomů řízených BRAF (19).
Další příklady modulátorů diferenciace zahrnují metabolit alfa-ketoglutarát (aKG), nezbytný kofaktor pro řadu enzymů modifikujících chromatin, o kterých bylo prokázáno, že se podílejí na stimulaci určitých stavů diferencovaných buněk. U karcinomu pankreatu stimuluje tumor supresor p53 produkci αKG a udržení diferencovanějšího buněčného stavu, zatímco prototypická ztráta funkce p53 vede ke snížení hladin αKG a následné dediferenciaci, která je spojena s maligní progresí (20).
U jedné formy rakoviny jater nevede mutace genu pro isocitrátdehydrogenázu (IDH1/2) k produkci diferenciace indukujícího αKG, ale spíše příbuzného „onkometabolitu“, D-2-hydroxygluterátu (D2HG), u kterého bylo prokázáno, že blokuje hepatocytární diferenciaci jaterních progenitorových buněk prostřednictvím D2HG-zprostředkované různé represe aquiescence hepatitidy. HNF4a.
D2HG zprostředkovaná suprese funkce HNF4a spouští proliferativní expanzi progenitorových buněk hepatocytů v játrech, které se stávají náchylnými k onkogenní transformaci po následné mutační aktivaci onkogenu KRAS, který řídí maligní progresi do cholangiokarcinomu jater (21). Mutant IDH1/2 a jeho onkometabolit D2HG také fungují u různých typů myeloidních a jiných solidních nádorů, kde D2HG inhibuje αKG-dependentní dioxygenázy potřebné pro děje metylace histonů a DNA, které zprostředkovávají změny ve struktuře chromatinu během vývojové diferenciace linie, čímž zmrazují nástup rakovinných buněk ve stavu progenitoru232 (32).
Dalším souvisejícím konceptem je „obejitá diferenciace“, ve které částečně nebo nediferencované progenitorové/kmenové buňky opouštějí buněčný cyklus a leží ladem v ochranných výklencích s potenciálem znovu iniciovat proliferativní expanzi ( 24 ), i když stále se selektivním tlakem narušit jejich naprogramovanou diferenciaci tím či oním způsobem.
Transdiferenciace
Koncept transdiferenciace je patology již dlouho uznáván ve formě tkáňové metaplazie, ve které buňky určitého diferencovaného fenotypu výrazně mění svou morfologii, aby se staly jasně rozpoznatelnými jako prvky jiné tkáně, jehož prominentním příkladem je Barrettův jícen, kde chronický zánět vrstevnatého dlaždicového epitelu vyvolává jednoduchý transteliferální epitel charakteristický pro transtezofag. střeva, čímž se usnadní následný vývoj adenokarcinomů spíše než spinocelulárních karcinomů očekávaných z tohoto dlaždicového epitelu (3).
Molekulární determinanty nyní odhalují mechanismy transdiferenciace u různých rakovin, a to jak pro případy, kdy je zřejmá metaplazie hrubé tkáně, tak pro jiné, kde je poněkud jemnější, jak ilustrují následující příklady.
Informativní případ transdiferenciace jako diskrétní události v tumorigenezi se týká pankreatického duktálního adenokarcinomu (PDAC), u kterého se jedna ze zapojených původních buněk, pankreatická acinární buňka, může během iniciace neoplastického vývoje transdiferencovat na fenotyp duktálních buněk. Dva TF – PTF1a a MIST1 – kontrolují specifikaci a udržování stavu diferencovaných acinárních buněk pankreatu prostřednictvím své exprese v kontextu samoudržujících se „feed-forward“ regulačních smyček (25).
Oba tyto TF jsou často downregulovány během neoplastického vývoje a maligní progrese lidského a myšího PDAC. Funkční genetické studie na myších a kultivovaných lidských PDAC buňkách ukázaly, že experimentálně vynucená exprese PTF1a narušuje KRAS-indukovanou transdiferenciaci a proliferaci a může také vynutit rediferenciaci již neoplastických buněk do klidového acinárního buněčného fenotypu (26).
Naopak suprese exprese PTF1a spouští metaplazii acinar-to-duct, konkrétně transdiferenciaci, a tím senzibilizuje duct-like buňky k onkogenní transformaci KRAS, což urychluje následný vývoj invazivního PDAC (27). Podobně vynucená exprese MIST1 v pankreatu exprimujícím KRAS také blokuje transdiferenciaci a zhoršuje iniciaci tumorigeneze pankreatu, která je jinak usnadněna tvorbou premaligních duct-like (PanIN) lézí, zatímco genetická delece MIST1 podporuje jejich tvorbu a iniciaci KRAS-řízené neoplastické progrese (28).
Ztráta exprese PTF1 nebo MIST1 během tumorigeneze je spojena se zvýšenou expresí dalšího vývojového regulačního TF, SOX9, který je normálně účinný ve specifikaci duktálních buněk (27, 28). Bylo také ukázáno, že vynucená upregulace SOX9, čímž se zabrání potřebě downregulace PTF1a, a MIST1 stimulují transdiferenciaci acinárních buněk na fenotyp duktálních buněk citlivý na neoplazii indukovanou KRAS (29), což implikuje SOX9 jako klíčový funkční efektor jejich downregulace v genezi lidské PDAC.
Tři TF, které regulují pankreatickou diferenciaci, tedy mohou být změněny různými způsoby, aby navodily transdiferencovaný stav, který v kontextu mutační aktivace KRAS usnadňuje onkogenní transformaci a zahájení tumorigeneze a maligní progrese.
Další členové SOX rodiny regulačních faktorů asociovaných s chromatinem jsou na jedné straně do značné míry spojeni jak se specifikací buněčného osudu a přepínáním linií ve vývoji (30), tak na druhé straně s několika fenotypy asociovanými s nádory (31). Další prominentní příklad transdiferenciace zprostředkované SOX zahrnuje mechanismus terapeutické rezistence u rakoviny prostaty.
V tomto případě je ztráta nádorových supresorů RB a p53 - jejichž absence je charakteristická pro neuroendokrinní nádory - v reakci na antiandrogenní terapii nezbytná, ale ne dostačující pro běžně pozorovanou transformaci dobře diferencovaných buněk rakoviny prostaty na buňky karcinomu, které napadly diferenciační linii s molekulárními a histologickými rysy neuroendokrinních buněk, které zejména neexprimují receptor. Kromě ztráty RB a p53 vyžaduje získaná rezistence na antiandrogenní terapii upregulovanou expresi SOX2, vývojového regulačního genu, u kterého bylo prokázáno, že pomáhá indukovat transdiferenciaci buněk adenokarcinomu reagujících na terapii na deriváty, které jsou ve stavu neuroendokrinních buněk refrakterním na terapii (32).
Třetí příklad také ukazuje transdiferenciaci jako strategii používanou karcinomovými buňkami, aby se vyhnuly eliminaci liniově specifickou terapií, v tomto případě s bazaliomy (BCC) kůže ošetřené farmakologickým inhibitorem onkogenní dráhy Hedgehog-Smoothened (HH/SMO), o které je známo, že řídí neoplastický růst těchto buněk ( 33).
Rakovinné buňky rezistentní vůči lékům přecházejí na vývojově příbuzný, ale odlišný typ buněk prostřednictvím širokých epigenetických posunů ve specifických chromatinových doménách a změněné dostupnosti dvou superenhancerů. Nově získaný fenotypový stav buněk BCC jim umožňuje udržovat expresi onkogenní signální dráhy WNT, která zase poskytuje nezávislost na signální dráze HH/SMO potlačované léky (34).
Jak se očekává z této transdiferenciace, transkriptom rakovinných buněk se posouvá z genového podpisu odrážejícího zapojenou buňku původu BCC, jmenovitě kmenové buňky z vyboulení vlasového folikulu, k podpisu indikujícímu bazální kmenové buňky osídlující BCC interfolikulární epidermis. Taková transdiferenciace umožňující rezistenci vůči lékům je stále častěji dokumentována u různých forem rakoviny (35).
Také se zdá, že plasticita vývojové linie převládá u hlavních podtypů plicního karcinomu, tj. h. u neuroendokrinních karcinomů [small cell lung cancer (SCLC)] a adenokarcinomů + spinocelulárních karcinomů [kolektivní nemalobuněčný karcinom plic (NSCLC)]. Sekvenování jednobuněčné RNA odhalilo pozoruhodně dynamickou a heterogenní konverzi mezi těmito podtypy, stejně jako jejich výrazné variace, během fází tumorigeneze plic, následné maligní progrese a odpovědi na terapii (36–38).
Proto spíše než jednoduchá konceptualizace čistého klonálního přechodu z jedné linie do druhé, tyto studie vykreslují mnohem složitější obraz dynamicky se vzájemně převádějících subpopulací rakovinných buněk, které vykazují rysy více vývojových linií a diferenciačních stádií, což je v tomto ohledu střízlivý pohled na terapeutické cílení na lidskou rakovinu plic na základě linie. Začínají být identifikovány regulační determinanty této dynamické fenotypové plasticity (37, 39, 40).
Shrnutí
Tři výše popsané třídy mechanismů zdůrazňují selektivní regulátory buněčné plasticity, které jsou – alespoň částečně – oddělitelné od jádrových onkogenních ovladačů a dalších charakteristických schopností. Kromě těchto příkladů existuje významný soubor důkazů spojujících mnoho forem rakoviny se zhoršenou diferenciací, což je doprovázeno získáním signatur transkriptomu a dalších fenotypů - například histologické morfologie - které jsou spojeny s progenitorovými nebo kmenovými buňkami pozorovanými v odpovídajících normálních tkáních. původu nebo v jiných vzdáleněji příbuzných buněčných typech a liniích (41 – 43).
Jako takové se tyto tři podtřídy fenotypové plasticity – dediferenciace zralých buněk zpět na progenitorové stavy, zastavená diferenciace za účelem zmrazení vyvíjejících se buněk ve stavech progenitorových/kmenových buněk a transdiferenciace na alternativní buněčné linie – zdají být účinné u několika typů rakoviny během primární tumorigeneze, maligní progrese a/nebo reakce na léčbu.
Existují však dvě koncepční úvahy. Za prvé, dediferenciace a zastavená diferenciace se pravděpodobně prolínají, protože jsou nerozeznatelné u mnoha typů nádorů, ve kterých je původní buňka – diferencovaná buňka nebo progenitorová/kmenová buňka – buď neznámá, nebo alternativně zapojená. Za druhé, získávání nebo udržování fenotypů progenitorových buněk a ztráta diferencovaných znaků je ve většině případů nepřesným odrazem normálního vývojového stadia, ponořením se do prostředí jiných charakteristických změn v rakovinné buňce, které nejsou přítomny v přirozeně se vyvíjejících buňkách.
Kromě toho další forma fenotypové plasticity zahrnuje buněčnou senescenci, o níž je obecněji pojednáno níže, přičemž rakovinné buňky indukované zdánlivě nevratným stárnutím jsou místo toho schopné uniknout a pokračovat v proliferativní expanzi (44). Konečně, stejně jako u jiných charakteristických schopností, buněčná plasticita není novým vynálezem nebo aberací rakovinných buněk, ale spíše poškozením latentních, ale aktivovatelných schopností, které různé normální buňky používají k podpoře homeostázy, opravy a regenerace (45).
Celkově tyto ilustrativní příklady podporují úvahu, že uvolnění buněčné plasticity pro umožnění různých forem narušené diferenciace představuje výraznou rozlišovací schopnost, která se liší regulací a buněčným fenotypem od dobře ověřených základních znaků rakoviny (obr. 2).
Epigenetické přeprogramování bez mutace
Umožňující vlastnost nestability a mutace genomu (DNA) je základní složkou rozvoje a patogeneze rakoviny. V současné době několik mezinárodních konsorcií katalogizuje mutace v celém genomu lidských rakovinných buněk, prakticky u každého typu lidské rakoviny, v různých fázích maligní progrese, včetně metastatických lézí, a během vývoje rezistence na adaptivní terapii. Jedním z výsledků je nyní rozšířené uznání, že mutace v genech, které organizují, modulují a udržují architekturu chromatinu, a tím regulují genovou expresi globálně, jsou stále častěji objevovány a funkčně spojeny s rakovinnými rysy (46–48).
Kromě toho existují argumenty pro další zdánlivě nezávislou formu přeprogramování genomu, která zahrnuje čistě epigeneticky regulované změny v genové expresi, takovou, kterou lze nazvat „nemutační epigenetické přeprogramování“ (obr. 3). Teze o bezmutační evoluci rakoviny a čistě epigenetickém programování charakteristických rakovinných fenotypů byla vznesena téměř před deseti lety (49) a je stále více diskutována (46, 50–52).
Obrázek 3

Podobně jako to, co se děje během embryogeneze a tkáňové diferenciace a homeostázy, hromadící se důkazy naznačují, že instrumentální genové regulační obvody a sítě v nádorech mohou být řízeny množstvím poškozených a kooptovaných mechanismů, které jsou nezávislé na nestabilitě genomu a genové mutaci. Ochranné známky grafiky rakoviny byly převzaty od Hanahana a Weinberga (2).
Samozřejmě, že koncept nemutační epigenetické regulace genové exprese je dobře zaveden jako centrální mechanismus zprostředkovávající embryonální vývoj, diferenciaci a organogenezi (53 – 55). U dospělých například dlouhodobá paměť zahrnuje změny v modifikaci genů a histonů, ve struktuře chromatinu a ve spouštění přepínačů genové exprese, které jsou v průběhu času stabilně udržovány pozitivními a negativními zpětnovazebními smyčkami (56, 57). Stále více důkazů podporuje myšlenku, že analogické epigenetické změny mohou přispět k získání charakteristických schopností během vývoje nádoru a maligní progrese. Pro podporu této hypotézy uvádíme níže několik příkladů.
Mikroenvironmentální mechanismy epigenetického přeprogramování
Pokud ne pouze prostřednictvím onkogenních mutací, jak se přeprogramuje genom rakovinných buněk? Rostoucí množství důkazů naznačuje, že aberantní fyzikální vlastnosti mikroprostředí nádoru mohou způsobit rozsáhlé změny v epigenomu, jejichž změny prospěšné pro fenotypový výběr schopností vlastností mohou vést ke klonálnímu růstu rakovinných buněk se zlepšenou schopností proliferativní expanzi.
Společným znakem nádorů (nebo oblastí v nádorech) je hypoxie v důsledku nedostatečné vaskularizace. Hypoxie například snižuje aktivitu TET demetyláz, což vede k významným změnám v methylomu, zejména hypermetylaci ( 58 ). Nedostatečná vaskularizace také pravděpodobně omezuje biologickou dostupnost kritických živin přenášených krví a například bylo prokázáno, že nedostatek živin mění kontrolu translace a následně zvyšuje maligní fenotyp buněk rakoviny prsu (59).
Přesvědčivým příkladem epigenetické regulace zprostředkované hypoxií je forma vždy fatálního dětského ependymomu. Stejně jako mnoho embryonálních a dětských nádorů tato forma postrádá opakující se mutace, zejména nedostatek řidičských mutací v onkogenech a nádorových supresorech. Spíše se ukázalo, že abnormální růst těchto rakovinných buněk je řízen programem regulace genů vyvolaným hypoxií (60, 61). Je pozoruhodné, že domnělá buňka původu této rakoviny sídlí v hypoxickém kompartmentu a pravděpodobně senzibilizuje buňky v něm, aby zahájila tumorigenezi prostřednictvím dosud neznámých kofaktorů.
Další přesvědčivý důkaz pro epigenetickou regulaci zprostředkovanou mikroprostředím se týká invazivní růstové schopnosti rakovinných buněk. Klasickým příkladem je reverzibilní indukce invazivity rakovinných buněk na okrajích mnoha solidních nádorů, řízená vývojovým regulačním programem známým jako přechod z epitelu na mezenchym (EMT; odkazy 62–64). Zejména bylo nedávno prokázáno, že hlavní regulátor EMT, ZEB1, indukuje expresi histonmethyltransferázy, SETD1B, která zase udržuje expresi ZEB1 v pozitivní zpětnovazební smyčce, která udržuje (invazivní) regulační stav EMT (65).
Předchozí studie podobně dokumentovala, že indukce EMT prostřednictvím upregulované exprese příbuzného TF, SNAIL1, způsobila výrazné změny v krajině chromatinu v důsledku indukce řady modifikátorů chromatinu, jejichž aktivita se ukázala jako nezbytná pro udržení fenotypového stavu (66). Kromě toho řada stavů a faktorů, kterým čelí rakovinné buňky na okrajích nádorů, včetně hypoxie a cytokinů vylučovaných stromálními buňkami, může zjevně indukovat EMT a tím invazivitu (67, 68).
Pozoruhodný příklad programování invazivity mikroprostředím, který údajně nesouvisí s programem EMT, zahrnuje autokrinní aktivaci neuronového signalizačního okruhu zahrnujícího sekretovaný glutamát a jeho receptor NMDAR (69, 70). Je pozoruhodné, že prototypická tuhost mnoha pevných nádorů, ztělesněná v rozsáhlých změnách extracelulární matrice (ECM), která obaluje buňky v nich, má hluboké důsledky pro invazivní a další fenotypové vlastnosti rakovinných buněk.
Ve srovnání s normální tkáňovou ECM, ze které nádory pocházejí, je nádorová ECM typicky charakterizována zvýšeným zesítěním a hustotou, enzymatickými modifikacemi a změněným molekulárním složením, které společně organizují, částečně prostřednictvím integrinových receptorů pro motivy ECM, signalizační a genové expresní sítě indukované ztuhlostí, které indukují invazivitu a další charakteristické rysy (71).
Kromě těchto regulačních mechanismů obdařených fyzickým nádorovým mikroprostředím může parakrinní signalizace, zahrnující rozpustné faktory uvolňované do extracelulárního prostředí různými buněčnými typy, které osídlují solidní nádory, také přispět k indukci několika morfologicky odlišných invazivních růstových programů (72), z nichž pouze jeden – nazývaný „mezenchymální“ – se zdá být zapojen do výše uvedeného mechanismu regulace eMT.
Epigenetická regulační heterogenita
Rostoucí znalostní základna zvyšuje uznání důležitosti intratumorální heterogenity při vytváření fenotypové diverzity, kde nejvhodnější buňky pro proliferativní expanzi a invazi přerostou své bratry, a jsou proto vybírány pro maligní progresi. Jeden aspekt této fenotypové heterogenity je jistě způsoben chronickou nebo epizodickou genomickou nestabilitou a výslednou genetickou heterogenitou v buňkách, které osídlují nádor.
Navíc je stále jasnější, že může existovat epigenetická heterogenita nezaložená na mutacích. Významným příkladem je linker histon H1.0, který je dynamicky exprimován a potlačován v subpopulacích rakovinných buněk v rámci řady typů nádorů, s následnou sekvestrací nebo dostupností domén o velikosti megabází [73]. Pozoruhodně bylo zjištěno, že populace rakovinných buněk s potlačenou H1.0 vykazuje vlastnosti podobné kmenům, zvýšenou schopnost iniciovat nádor a asociaci se špatnou prognózou u pacientů.
Další příklad epigeneticky regulované plasticity byl popsán u lidských orálních spinocelulárních karcinomů (SCC), kde rakovinné buňky na invazivních okrajích přijmou částečný stav EMT (p-EMT), který postrádá výše uvedené mezenchymální TF, ale exprimuje jiné geny definující EMT, které nejsou exprimovány v centrálním jádru nádorů (74).
Buňky p-EMT zjevně nepředstavují klonální kompartmentalizaci mutačně změněných buněk: kultury primárních rakovinných buněk odvozených od nádoru obsahují dynamické směsi buněk p-EMT hi a p-EMT lo a když byly buňky p-EMT hi/lo purifikovány a kultivovány FACS, obě se vrátily do smíšených populací p-EMT hi a p-EMT lo během 4 dnů. Ačkoli parakrinní signály ze sousedního stromatu by mohly být považovány za deterministické pro stav p-EMT hi, stabilní přítomnost a regenerace dvou epigenetických stavů v kultuře svědčí pro vnitřní mechanismus rakovinné buňky. Tento závěr je podpořen zejména analýzou 198 buněčných linií reprezentujících 22 typů rakoviny, včetně SCC, kde 12 stabilně heterogenních epigenetických stavů (včetně p-EMT v SCC) bylo různě detekováno v modelech buněčných linií, stejně jako jejich související primární nádory (75).
Opět, heterogenní fenotypové stavy nemohly být spojeny s detekovatelnými genetickými rozdíly a v několika případech se ukázalo, že FACS-tříděné buňky konkrétního stavu se po kultivaci dynamicky znovu ekvilibrují, čímž se rekapituluje stabilní rovnováha mezi heterogenními stavy pozorovanými v původních buněčných liniích.
Kromě toho technologie pro profilování různých atributů v celém genomu – kromě sekvence DNA a jejích mutačních variací – osvětlují vlivné prvky anotace a organizace genomu rakovinných buněk, které korelují s prognózou pacienta a stále více s charakteristickými schopnostmi (76 – 78). Epigenomickou heterogenitu odhalují stále výkonnější technologie profilování metylace DNA v celém genomu (79, 80), modifikace histonů (81), dostupnosti chromatinu (82) a post-transkripční modifikace a translace RNA (83, 84).
Výzvou s ohledem na zde uvažovaný postulát bude určit, které epigenomické modifikace u určitých typů rakoviny (i) mají regulační význam a (ii) jsou reprezentativní pro čistě nemutační přeprogramování, na rozdíl od mutací řízené a tedy genomově vysvětlitelné nestability.
Epigenetická regulace typů stromálních buněk obývajících nádorové mikroprostředí
Obecně se předpokládá, že přídatné buňky v mikroprostředí nádoru, které funkčně přispívají k získání charakteristických schopností, netrpí genetickou nestabilitou a mutačním přeprogramováním za účelem zvýšení jejich aktivit podporujících nádor; spíše se dochází k závěru, že tyto buňky – s rakovinou asociované fibroblasty, vrozené imunitní buňky a endoteliální buňky a pericyty nádorové vaskulatury – jsou epigeneticky přeprogramovány po svém náboru rozpustnými a fyzikálními faktory, které definují mikroprostředí solidního nádoru (2, 85).
Očekává se, že technologie multi-omického profilování, které se v současnosti používají na rakovinné buňky, budou stále více používány ke studiu přídatných (stromálních) buněk v nádorech, aby se objasnilo, jak jsou normální buňky poškozeny, aby funkčně podpořily vývoj a progresi nádoru. Například nedávná studie (86) naznačuje, že takové přeprogramování může zahrnovat modifikace epigenomu, navíc k indukční výměně cytokinů, chemokinů a růstových faktorů, které mění intracelulární signální sítě ve všech těchto typech buněk:
Když byly myší modely s plicními metastázami ošetřeny kombinací inhibitoru DNA metyltransferázy (5-azacytidin) a inhibitoru modifikace histonů (HDAC), bylo zjištěno, že infiltrující myeloidní buňky přešly z nezralého (nádor podporujícího) progenitorového stavu do buněk připomínajících zralé intersticiální (nádorové antagonizující) schopnosti, na rozdíl od svých požadovaných nádorových protějšků, které nebyly schopny podporovat typické nádorové protějšky. pro účinnou metastatickou kolonizaci (86). Je možné, že multiomické profilování a farmakologické poruchy poslouží k objasnění přeprogramovaného epigenetického stavu v takových myeloidních buňkách, stejně jako v jiných charakteristických přídatných typech buněk, které osídlují mikroprostředí nádoru.
Shrnutí
Dohromady tyto ilustrativní snímky podporují tezi, že epigenetické přeprogramování bez mutace bude akceptováno jako skutečně umožňující vlastnost, která slouží k usnadnění získání charakteristických schopností (obr. 3), odlišných od nestability a mutace genomové DNA. Zejména lze očekávat, že nemutační epigenetické přeprogramování se ukáže jako nedílné pro umožnění předběžné nové rozlišovací schopnosti fenotypové plasticity diskutované výše, zejména jako hnací síla v dynamické transkriptomické heterogenitě, která je stále lépe dokumentována v TME maligních rakovinných buněk. Pokrok v jednobuněčných multi-omických profilovacích technologiích osvětlí příslušné příspěvky a souhru mezi mutací řízenou a nemutací řízenou epigenetickou regulací ve vývoji nádorů během maligní progrese a metastáz.
Polymorfní mikrobiomy
Dalekosáhlá hranice v biomedicíně se otevírá osvětlováním rozmanitosti a variability množství mikroorganismů, souhrnně označovaných jako mikrobiota, které se symbioticky spojují s tělesnými bariérovými tkáněmi vystavenými vnějšímu prostředí - zejména epidermis a vnitřní sliznici gastrointestinálního traktu, stejně jako plíce, prsa a urogenitální systém.
Stále více se uznává, že ekosystémy vytvořené rezidentními bakteriemi a houbami – mikrobiomy – mají hluboký vliv na zdraví a nemoci ( 87 ), což je uvědomění poháněné schopností prověřovat populace mikrobiálních druhů pomocí sekvenování nové generace a bioinformatických technologií. U rakoviny jsou důkazy stále přesvědčivější, že polymorfní variabilita v mikrobiomech mezi jednotlivci v populaci může mít hluboký vliv na fenotypy rakoviny (88, 89).
Asociační studie lidských a experimentálních manipulací na myších modelech rakoviny odhalují určité mikroorganismy, primárně, ale ne výhradně bakterie, které mohou mít buď ochranné nebo škodlivé účinky na vývoj rakoviny, maligní progresi a odpověď na terapii. To platí i pro globální komplexnost a složení tkáňového mikrobiomu jako celku. Zatímco střevní mikrobiom byl průkopníkem této nové hranice, několik tkání a orgánů má asociované mikrobiomy, které vykazují charakteristické rysy související s populační dynamikou a rozmanitostí mikrobiálních druhů a poddruhů.
Toto rostoucí uznání důležitosti polymorfně proměnných mikrobiomů ve zdraví a nemoci vyvolává otázku: Je mikrobiom zřetelnou umožňující vlastností, která široce ovlivňuje, jak pozitivně, tak negativně, získávání charakteristických schopností pro rakovinu? Tuto možnost zvažuji níže a ilustruji důkazy pro některé z prominentních tkáňových mikrobiomů zapojených do rysů rakoviny (obr. 4), počínaje nejvýraznějším a zřejmě nejpůsobivějším mikrobiomem, mikrobiomem střevního traktu.
Obrázek 4
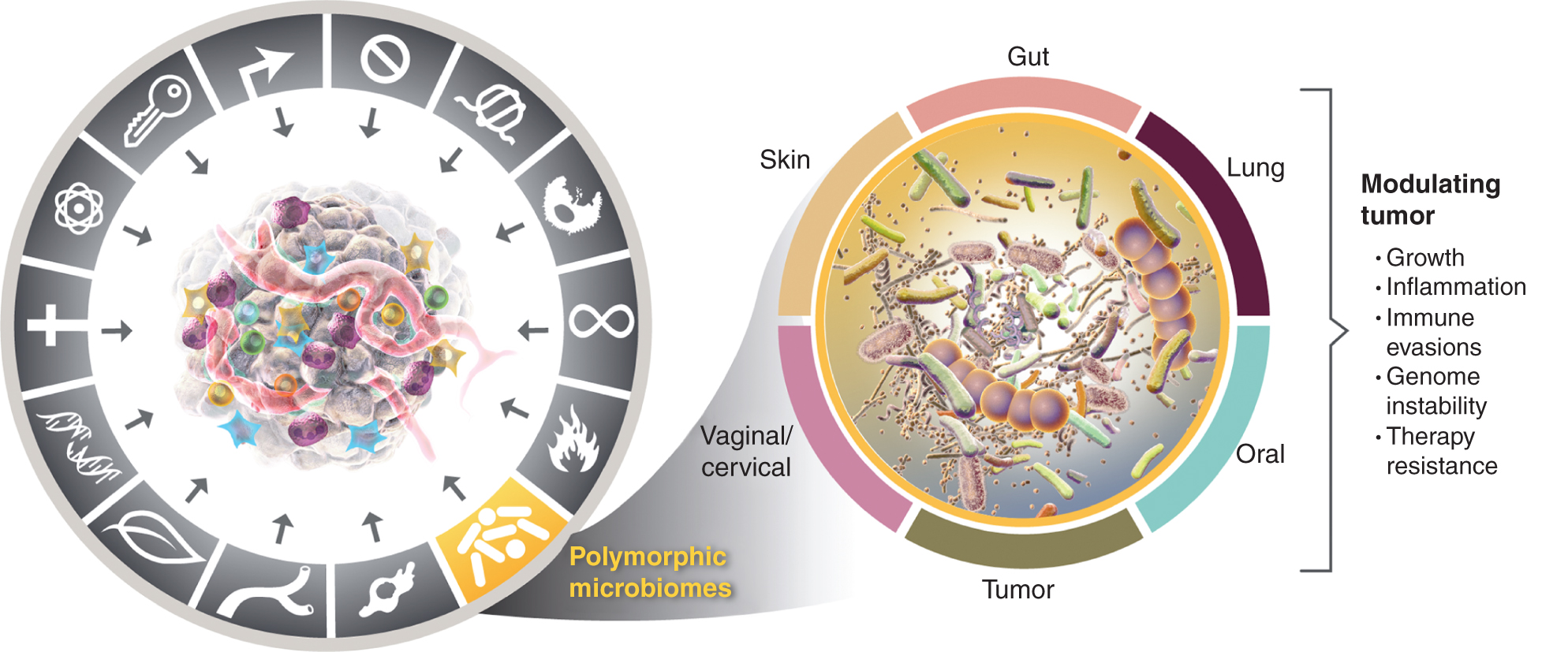
Vlevo, zatímco se aktivační vlastnosti zánětu podporujícího nádor a genomová nestabilita a mutace překrývají, existuje stále více důvodů k závěru, že polymorfní mikrobiomy umístěné u jednoho jedince ve srovnání s druhým v tlustém střevě, v jiných sliznicích a přidružených orgánech nebo v nádorech samotných mohou ovlivňovat řadu charakteristických schopností různými způsoby – buď indukcí nebo inhibicí –, a proto mohou být kvazi-instrukčním nástrojem progrese a hádanky. roste reaguje na terapii. Pravda, na modulaci nádorových fenotypů se podílí více tkáňových mikrobiomů. Kromě široce studovaného střevního mikrobiomu se na modulaci získávání – pozitivních i negativních – charakteristických schopností přítomných u určitých typů nádorů podílejí další charakteristické tkáňové mikrobiomy a také mikrobiom nádoru. Ochranné známky grafiky rakoviny byly převzaty od Hanahana a Weinberga (2).
Mnohonásobné modulační účinky střevního mikrobiomu
Již dlouho je známo, že střevní mikrobiom je zásadní pro funkci tlustého střeva (tlustého střeva) při štěpení a importu živin do těla v rámci metabolické homeostázy a že narušení mikrobiálních populací – dysbióza – v tlustém střevě může způsobit řadu fyziologických onemocnění (87). To zahrnuje podezření, že náchylnost, vývoj a patogeneze rakoviny tlustého střeva je ovlivněna střevním mikrobiomem. V posledních letech přesvědčivé funkční studie s použitím fekálních transplantací od pacientů s nádorem tlustého střeva a myší do myší příjemců predisponovaných k rozvoji rakoviny tlustého střeva stanovily princip: existují mikrobiomy chránící před rakovinou a podporující nádory zahrnující specifické bakteriální druhy, které mohou modulovat výskyt a patogenezi nádorů tlustého střeva (90).
Mechanismy, kterými mikrobiota uděluje tyto modulační role, jsou stále objasňovány, ale dva obecné účinky jsou stále více dobře zavedeny pro mikrobiomy podporující nádory a v některých případech pro specifické bakteriální druhy podporující nádor. Prvním účinkem je mutageneze epitelu tlustého střeva jako výsledek produkce bakteriálních toxinů a dalších molekul, které buď přímo poškozují DNA, nebo narušují systémy udržující genomickou integritu nebo jinak stresují buňky, čímž nepřímo ovlivňují věrnost replikace a opravy DNA. Typickým příkladem je E. coli, která nese lokus PKS, který prokazatelně mutagenizuje lidský genom a podílí se na přenosu mutací umožňujících znaménko (91).
Dále bylo popsáno, že se bakterie vážou na povrch epiteliálních buněk tlustého střeva a produkují mimetika ligandů, která stimulují proliferaci epitelu, což přispívá k charakteristické proliferativní signalizační schopnosti v neoplastických buňkách (88). Dalším mechanismem, kterým specifické typy bakterií podporují vývoj nádoru, jsou bakterie produkující butyrát, jejichž četnost je zvýšena u pacientů s kolorektálním karcinomem (92).
Produkce metabolitu butyrátu má komplexní fyziologické účinky, včetně indukce senescentních epiteliálních a fibroblastových buněk. U myšího modelu karcinogeneze tlustého střeva kolonizovaného bakteriemi produkujícími butyrát se vyvinulo více nádorů než u myší bez takových bakterií; Souvislost mezi stárnutím vyvolaným butyrátem a zvýšenou tumorigenezí tlustého střeva byla prokázána použitím senolytického léku, který zabíjí senescentní buňky a narušuje růst nádoru (92).
Bakterie produkovaný butyrát má navíc pleiotropní a paradoxní účinky na diferencované buňky ve srovnání s nediferencovanými (kmenovými) buňkami v epitelu tlustého střeva v podmínkách, kdy je narušena střevní bariéra (dysbióza) a bakterie jsou invazivní, ovlivňující například buněčnou energii a metabolismus, modifikaci histonů, progresi buněčného cyklu a (imunitní podporu podporující zánět imunitního systému93) adaptaci imunitního systému.
Široké působení polymorfních mikrobiomů skutečně zahrnuje modulaci adaptivního a vrozeného imunitního systému prostřednictvím různých cest, včetně produkce „imunomodulačních“ faktorů bakteriemi, které aktivují senzory poškození na epiteliálních nebo rezidentních imunitních buňkách, což vede k expresi různorodého repertoáru chemokinů a cytokinů, které mohou utvářet hojnost a drenáž imunitních buněk a drenážní vlastnosti imunitních buněk tlustého střeva. uzly.
Kromě toho mohou některé bakterie narušit jak ochranný biofilm, tak hlen lemující epitel tlustého střeva a narušit těsná spojení epiteliální buňka-buňka, která společně udržují integritu fyzické bariéry, která normálně kompartmentalizuje střevní mikrobiom. Po invazi do stromatu mohou bakterie spustit jak vrozené, tak adaptivní imunitní reakce tím, že vyvolají sekreci řady cytokinů a chemokinů. Jedním z projevů může být vytvoření nádor podporujícího nebo nádor antagonizujícího imunitního mikroprostředí, které následně chrání nebo usnadňuje tumorigenezi a maligní progresi.
V souladu s tím může být modulace vzájemně provázaných parametrů (i) indukce (vrozeného) zánětu podporujícího nádor a (ii) úniku z (adaptivní) imunitní destrukce charakteristickými mikrobiomy u jednotlivých pacientů spojena nejen s prognózou, ale také s odpovědí nebo rezistencí na imunoterapie s inhibitory imunitního kontrolního bodu a dalšími terapeutickými modalitami (Jedním z projevů může být tvorba nádorů podporující mikrovibraci nebo tvorba imunitních stimulátorů vyskytují Chrání nebo usnadňují vývoj nádoru a maligní progresi.
V souladu s tím může být modulace vzájemně provázaných parametrů (i) indukce (vrozeného) zánětu podporujícího nádor a (ii) úniku z (adaptivní) imunitní destrukce charakteristickými mikrobiomy u jednotlivých pacientů spojena nejen s prognózou, ale také s odpovědí nebo rezistencí na imunoterapie s inhibitory imunitního kontrolního bodu a dalšími terapeutickými modalitami (Jedním z projevů může být tvorba nádorů podporující mikrovibraci nebo tvorba imunitních stimulátorů dojít k ochraně nebo usnadnění vývoje nádoru a maligní progrese).
V souladu s tím může být modulace vzájemně provázaných parametrů (i) indukce (vrozeného) zánětu podporujícího nádor a (ii) úniku z (adaptivní) imunitní destrukce pomocí odlišných mikrobiomů u jednotlivých pacientů spojena nejen s prognózou, ale také s odpovědí nebo rezistencí na imunoterapie s inhibitory imunitního kontrolního bodu a dalšími terapeutickými modalitami (89, 94–96). Předběžný důkaz konceptu pochází z nedávných studií prokazujících obnovenou účinnost imunoterapie po transplantacích fekální mikroflóry od pacientů, kteří reagují na léčbu, pacientům s melanomem, který progredoval během předchozí léčby blokádou kontrolních bodů imunity (97, 98).
Molekulární mechanismy, jimiž odlišné a variabilní složky střevního mikrobiomu systémově modulují aktivitu adaptivního imunitního systému, zůstávají přetrvávající záhadou, ať už posílením protinádorových imunitních reakcí vyvolaných blokádou imunitního kontrolního bodu, nebo spíše indukcí systémové nebo lokální (intratumorální) imunosuprese. Nedávná studie vrhla světlo: některé kmeny Enterococcus (a další bakterie) exprimují peptidoglykanovou hydrolyázu zvanou SagA, která uvolňuje mukopeptidy z bakteriální stěny, které pak mohou systémově cirkulovat a aktivovat receptor NOD2, což zase zvyšuje odpovědi T-buněk a účinnost kontrolní imunoterapie (99).
Jsou identifikovány a funkčně hodnoceny další imunoregulační molekuly produkované specifickými bakteriálními poddruhy, včetně bakteriálně produkovaného inosinu, metabolitu omezujícího rychlost aktivity T buněk (100). Tyto a další příklady začínají vymezovat molekulární mechanismy, kterými polymorfní mikrobiomy nepřímo a systémově modulují nádorovou imunobiologii, nad rámec imunitních odpovědí, které následují po přímých fyzikálních interakcích bakterií s imunitním systémem (101, 102).
Kromě kauzálních souvislostí s rakovinou tlustého střeva a melanomem je prokázaná schopnost střevního mikrobiomu vyvolat expresi imunomodulačních chemokinů a cytokinů, které vstupují do systémové cirkulace, také zjevně schopna ovlivnit patogenezi rakoviny a odpověď na terapie v jiných orgánech těla (94, 95).
Ilustrativní příklad se týká vývoje cholangiokarcinomů v játrech: střevní dysbióza umožňuje vstup a transport bakterií a bakteriálních produktů přes portální žílu do jater, kde je TLR4 exprimovaný na hepatocytech spuštěn k indukci exprese chemokinu CXCL1, který rekrutuje CXCR2-exprimující granulocytární myeloidní buňky (gMD03kde) a pravděpodobně slouží k přenosu jiných přirozených imunitních buněk (gMD03kde) výrazné schopnosti (85). Střevní mikrobiom jako takový je jasně implikován jako podpůrná vlastnost, která může alternativně usnadňovat nebo chránit před více rakovinami.
Za střevem: Implementace odlišných mikrobiomů do jiných bariérových tkání
Téměř všechny tkáně a orgány, které jsou přímo či nepřímo vystaveny vnějšímu prostředí, jsou zároveň úložištěm komenzálních mikroorganismů ( 104 ). Na rozdíl od střeva, kde je symbiotická role mikrobiomu v metabolismu dobře známa, se normální a patogenní role rezidentní mikrobioty v těchto různorodých lokalitách stále objevují.
Existují zjevné orgánově/tkáňově specifické rozdíly v konstituci souvisejících mikrobiomů při homeostáze, stárnutí a rakovině, s překrývajícími se a odlišnými druhy a frekvencemi oproti těm v tlustém střevě (104, 105). Kromě toho asociační studie poskytují stále více důkazů o tom, že lokální tkáňové mikrobiomy antagonizující/protektivní versus nádory podporující, podobné střevnímu mikrobiomu, mohou modulovat náchylnost a patogenezi k lidským rakovinám vznikajícím v jejich přidružených orgánech (106–109).
Vliv intratumorální mikrobioty?
A konečně, patologové již dlouho uznali, že bakterie lze detekovat v pevných nádorech, což je pozorování, které bylo nyní podloženo sofistikovanými technologiemi profilování. Například ve studii 1 526 nádorů zahrnujících sedm lidských typů rakoviny (kost, mozek, prsa, plíce, melanom, vaječníky a slinivka) se každý typ vyznačoval charakteristickým mikrobiomem, který se z velké části nachází v rakovinných buňkách a imunitních buňkách. U každého typu nádoru byly prokázány variace v mikrobiomu nádoru a došlo k závěru, že souvisejí s klinicko-patologickými rysy (110).
Mikrobiota byla podobně detekována v de novo geneticky upravených myších modelech rakoviny plic a slinivky břišní a jejich nepřítomnost u bezmikrobních myší a/nebo jejich zrušení antibiotiky může narušit tumorigenezi, funkčně implikující nádorový mikrobiom jako prekurzor zánětu podporujícího nádor a maligní progresi (1211, 11).
Asociační studie u lidského duktálního adenokarcinomu slinivky břišní a funkční testy prostřednictvím fekální transplantace myším s nádorem ukázaly, že variace v nádorovém mikrobiomu – a souvisejícím střevním mikrobiomu – modulují fenotypy imunitního systému a přežití (113). Důležitou výzvou pro budoucnost bude rozšířit tyto důsledky na další typy nádorů a oddělit potenciálně oddělitelné příspěvky konstituce a variace v mikrobiomu nádoru od příspěvků střevního mikrobiomu (a místní tkáně původu), možná identifikací specifických mikrobiálních druhů, které jsou funkčně vlivné na jednom místě nebo jiném.
Shrnutí
Zajímavé otázky budoucnosti zahrnují, zda mikrobiota sídlící v různých tkáních nebo osídlující počínající novotvary mají schopnost přispět nebo narušit získání dalších charakteristických schopností mimo imunomodulaci a genomové mutace, a tím ovlivnit vývoj a progresi nádoru. Existují důkazy, že určité bakteriální druhy mohou přímo stimulovat charakteristický znak proliferativní signalizace, například v epitelu tlustého střeva (88), a mohou modulovat supresi růstu změnou supresorové aktivity nádoru v různých částech střeva (114), zatímco přímé účinky na jiné charakteristické schopnosti, jako je zamezení buněčné smrti, spouštění angiogeneze a stimulace těchto obecných invazí a mnohočetných pozorování rakoviny, zůstávají i nadále rakovinou.
Bez ohledu na to existují stále přesvědčivější argumenty, že polymorfní variace v mikrobiomech střeva a dalších orgánů představuje výrazný aktivační rys pro získání rozlišovacích dovedností (obr. 4), i když se překrývá a doplňuje s genomovou nestabilitou a mutací a zánětem podporujícím nádor.
Senescentní buňky
Buněčné stárnutí je typicky nevratná forma proliferativní zástavy, která se pravděpodobně vyvinula jako ochranný mechanismus pro udržení tkáňové homeostázy, zdánlivě jako doplňkový mechanismus k programované buněčné smrti, která slouží k inaktivaci a v patřičnou dobu k odstranění nemocných, dysfunkčních nebo jinak nepotřebných buněk. Kromě zastavení cyklu buněčného dělení vyvolává program stárnutí změny v morfologii a metabolismu buněk a nejhlubší aktivaci sekrečního fenotypu spojeného se stárnutím (SASP), který zahrnuje uvolnění velkého množství bioaktivních proteinů, včetně chemokinů.
Cytokiny a proteázy, jejichž identita závisí na typu buňky a tkáně, ze kterých senescentní buňka pochází (115–117). Senescence může být v buňkách vyvolána různými podmínkami, včetně mikroenvironmentálních stresů, jako je hladovění živin a poškození DNA, stejně jako poškození organel a buněčné infrastruktury a nerovnováha v buněčných signálních sítích (115, 117), ke všemu došlo v souvislosti s pozorovaným zvýšením frekvence senescentních buněk v různých orgánech během stárnutí (19,19).
Stárnutí buněk bylo dlouho považováno za ochranný mechanismus proti neoplazii, který způsobuje stárnutí rakovinných buněk ( 120 ). Většina výše zmíněných iniciátorů programu senescence je spojena s malignitou, zejména poškozením DNA v důsledku aberantní hyperproliferace, tzv. onkogenem indukovaná senescence v důsledku hyperaktivované signalizace a terapií indukovaná senescence v důsledku buněčného a genomového poškození způsobeného chemoterapií a radioterapií.
Opravdu existují dobře zavedené příklady ochranných přínosů stárnutí při omezení maligní progrese (118, 119). Naopak, rostoucí množství důkazů ukazuje pravý opak: v určitých kontextech senescentní buňky diferencovaně stimulují vývoj nádoru a maligní progresi (119, 121).
V přehledné případové studii byly senescentní buňky u stárnoucích myší farmakologicky ablatovány, konkrétně deplecí senescentních buněk, které charakteristicky exprimují inhibitor buněčného cyklu p16 – INK4a: kromě oddálení několika symptomů souvisejících s věkem to vedlo k depleci senescentních buněk u stárnoucích myší se sníženým výskytem spontánní tumorigeneze a úmrtí souvisejícího s rakovinou (122).
Předpokládá se, že hlavním mechanismem, kterým senescentní buňky podporují nádorové fenotypy, je SASP, u kterého bylo prokázáno, že je schopen zprostředkovat signální molekuly (a proteázy, které se aktivují a/nebo deaktivují) parakrinním způsobem ke zprostředkování typických schopností. V různých experimentálních systémech se tedy ukázalo, že senescentní rakovinné buňky přispívají různými způsoby k proliferativní signalizaci, vyhýbají se apoptóze, indukují angiogenezi, stimulují invazi a metastázy a potlačují nádorovou imunitu (116, 118, 120, 121).
Ještě další aspekt účinků senescentních rakovinných buněk na rakovinové fenotypy zahrnuje přechodné, reverzibilní stavy senescentních buněk, kdy senescentní rakovinné buňky mohou uniknout svému neproliferativnímu stavu exprimujícímu SASP a obnovit buněčnou proliferaci a projev souvisejících schopností plně životaschopných onkogenových buněk (44).
Takové přechodné stárnutí je nejlépe dokumentováno v případech rezistence na terapii (44), která představuje formu klidu, která se vyhýbá terapeutickému zacílení na proliferující rakovinné buňky, ale může se ukázat jako širší účinnost v jiných fázích vývoje nádoru, maligní progrese a metastáz.
Kromě toho, schopnosti senescentních buněk podporovat charakteristické znaky nejsou omezeny na senescentní rakovinné buňky. Bylo prokázáno, že fibroblasty spojené s rakovinou (CAF) senescují v nádorech, což vede ke vzniku senescentních CAF, u kterých bylo prokázáno, že podporují nádory tím, že udělují charakteristické schopnosti rakovinným buňkám v TME (115, 116, 121).
Kromě toho senescentní fibroblasty v normálních tkáních, tvořené zčásti přirozeným stárnutím nebo vnějšími vlivy, se podobně podílejí na remodelaci tkáňových mikroprostředí prostřednictvím svých SASP, aby poskytovaly parakrinní podporu pro místní invazi (takzvané „efekty pole“) a vzdálené metastázy (116) novotvarů vyvíjejících se poblíž.
Kromě toho se ukázalo, že senescentní fibroblasty ve stárnoucí pokožce rekrutují – prostřednictvím svých SASP – vrozené imunitní buňky, které jsou imunosupresivní vůči adaptivním protinádorovým imunitním odpovědím zakotveným CD8 T buňkami a stimulují růst kožního nádoru (123), přičemž tento účinek pravděpodobně odráží parakrinní příspěvky takových vrozených imunitních buněk (myeloidních buněk, neutrofilů a makrofágů) k dalším charakteristickým schopnostem.
I když je to méně dobře prokázané, zdá se pravděpodobné, že další hojné stromální buňky obývající specifická nádorová mikroprostředí podstoupí senescenci, čímž budou modulovat charakteristiky rakoviny a výsledné nádorové fenotypy. Terapií indukované senescentní nádorové endoteliální buňky mohou například zvýšit proliferaci, invazi a metastázy v modelech rakoviny prsu (124, 125).
Takové důkazy jistě vyžadují zkoumání jiných typů nádorů, aby se vyhodnotilo celkové stárnutí fibroblastů, endoteliálních buněk a dalších stromálních buněk jako hnací síly ve vývoji nádoru. V současné době jsou také nejasné regulační mechanismy a funkční determinanty, kterými konkrétní senescentní buněčný typ v konkrétním TME vyvolává SASP podporující nádor versus nádor antagonizující SASP, které zjevně mohou být indukovány alternativně ve stejném senescentním buněčném typu, možná různými iniciátory, když jsou ponořeny do charakteristických fyziologických a neoplastických mikroprostředí.
Shrnutí
Koncepce, že nádory sestávají z geneticky transformovaných rakovinných buněk, které interagují a mají prospěch z rekrutovaných a epigeneticky/fenotypicky poškozených přídatných (stromálních) buněk, byla stanovena jako klíčová pro patogenezi rakoviny. Úvahy diskutované výše a popsané v přehledech a zprávách citovaných zde (a jinde) přesvědčivě argumentují, že senescentní buňky (bez ohledu na buněčný původ) by měly být zváženy pro zařazení do seznamu funkčně významných buněk v mikroprostředí nádoru (obr. 5). Proto by se při hledání hloubkových znalostí mechanismů rakoviny měly brát v úvahu senescentní buňky. Kromě toho uznání jejich důležitosti motivuje sekundární cíl terapeutického cílení senescentních buněk podporujících nádor všech konstitucí, ať už prostřednictvím farmakologické nebo imunologické ablace nebo přeprogramováním SASP na varianty antagonizující nádor (115, 121, 126).
Obrázek 5

Heterogenní podtypy rakovinných buněk a typy a podtypy stromálních buněk jsou funkčně integrovány do projevů nádorů jako nelegální orgány. Stále více důkazů naznačuje, že senescentní buněčné deriváty mnoha z těchto buněčných složek TME a jejich variabilní SASP se podílejí na modulaci charakteristických schopností a výsledných nádorových fenotypů. Ochranné známky grafiky rakoviny byly převzaty od Hanahana a Weinberga (2).
Závěrečné poznámky
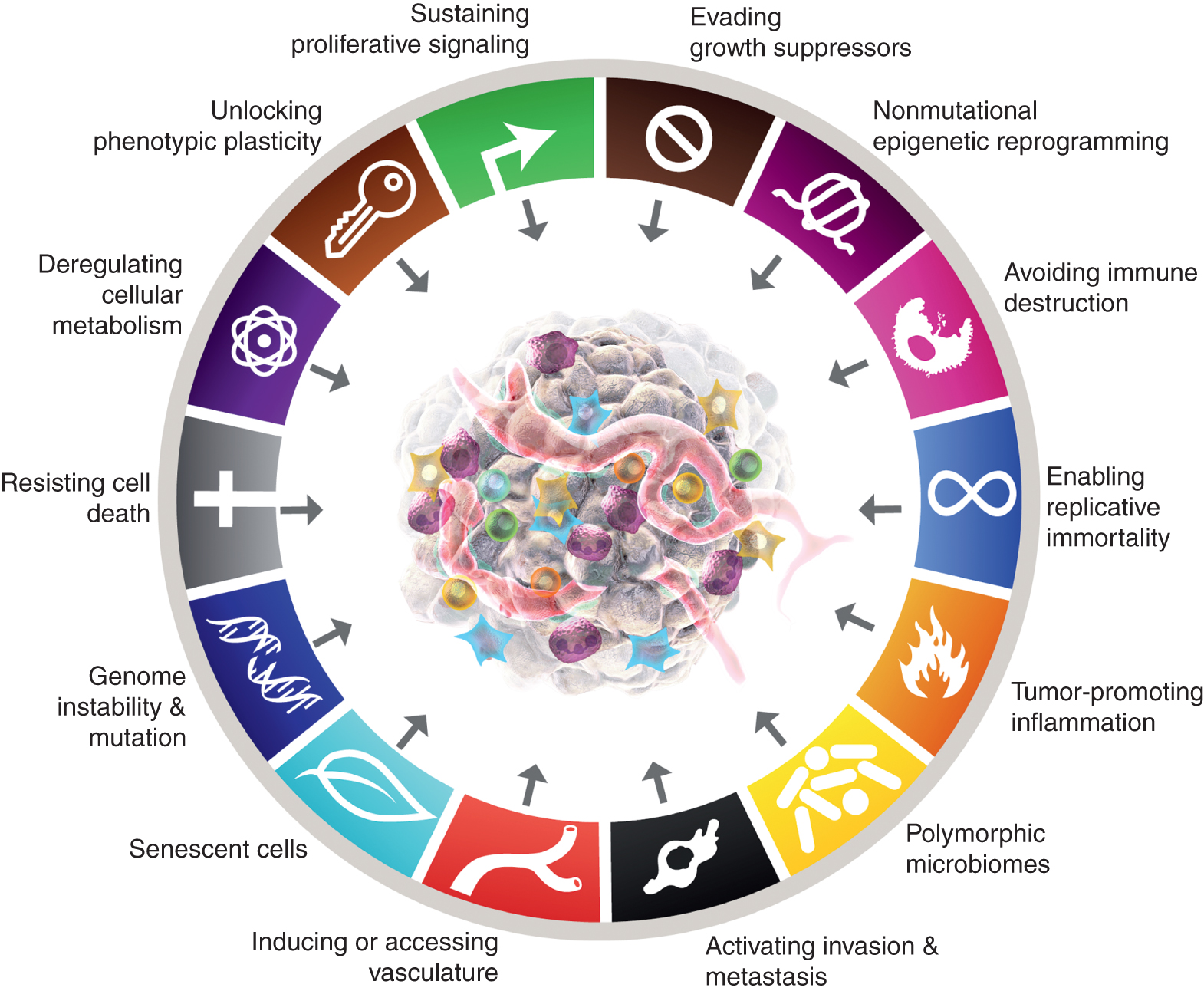
I když se ukázalo, že osm charakteristických znaků rakoviny a jejich dva podpůrné rysy mají trvalou heuristickou hodnotu v konceptualizaci rakoviny, výše uvedené úvahy naznačují, že mohou existovat nové aspekty určité obecnosti, a tedy důležité pro úplnější pochopení složitosti, mechanismů a projevů nemoci. Aplikací metriky rozeznatelné, ne-li úplné nezávislosti na 10 základních atributech, je sporné, že tyto čtyři parametry – po další validaci a zobecnění nad rámec prezentovaných případových studií – mohou být dobře integrovány do charakteristických znaků schématu rakoviny (obr. 6).
Buněčná plasticita by proto mohla být přidána na seznam prominentních schopností. Zatímco osmé jádro a tato nová schopnost jsou koncepčně odlišitelné svou definicí jako charakteristickými znaky, aspekty jejich regulace jsou u některých a možná i mnoha druhů rakoviny alespoň částečně spojeny. Například mnohočetné charakteristické znaky jsou koordinovaně modulovány kanonickými onkogenními ovladači u některých typů nádorů, včetně
- (I) KRAS ( https://cancer.sanger.ac.uk/cosmic/census-page/KRAS ),
- (II) MYC ( https://cancer.sanger.ac.uk/cosmic/census-page/MYC ),
- (III) NOTCH ( https://cancer.sanger.ac.uk/cosmic/census-page/NOTCH1 ; Ref. 127) und
- (IV) TP53 ( https://cancer.sanger.ac.uk/cosmic/census-page/TP53 )
Obrázek 6
Jsou zobrazeny kanonické a očekávané nové přírůstky do „Hallmarks of Cancer“. Tento dokument nastoluje možnost, s cílem podnítit debatu, diskusi a experimentální zpracování, že některé nebo všechny ze čtyř nových parametrů budou uznány jako generické pro mnohočetné formy lidské rakoviny, a tudíž vhodné pro integraci do základní konceptualizace charakteristických znaků rakoviny. Ochranné známky grafiky rakoviny byly převzaty od Hanahana a Weinberga (2).
Kromě přidání buněčné plasticity do seznamu lze do orgánových/tkáňových mikrobiomů integrovat nemutační epigenetické přeprogramování a polymorfní variace jako mechanické determinanty – umožňující vlastnosti – jejichž prostřednictvím se získávají výrazné schopnosti spolu se zánětem podporujícím nádor (sám částečně propojený s mikrobiomem), nad rámec mutací a dalších aberací, které se projevují onkogenními faktory.
Konečně, senescentní buňky různého původu – včetně rakovinných buněk a různých stromálních buněk – které funkčně přispívají k rozvoji a maligní progresi rakoviny, i když výrazně odlišným způsobem od buněk jejich nesenescentních bratří, mohou být zahrnuty jako generické složky TME. Stručně řečeno, předpokládá se, že nasazení těchto předběžných „experimentálních balónků“ podnítí debatu, diskusi a další experimentální zkoumání v komunitě pro výzkum rakoviny ohledně definujících koncepčních parametrů biologie, genetiky a patogeneze rakoviny.
Reference
- Hanahan D , Weinberg RA . The hallmarks of cancer. Cell 2000;100:57–70.
- Hanahan D , Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Yuan S , Norgard RJ , Stanger BZ . Cellular plasticity in cancer. Cancer Discov 2019;9:837–51.
- Barker N , Ridgway RA , van Es JH , van de Wetering M , Begthel H , van den Born M et al . Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608–11.
- Perekatt AO , Shah PP , Cheung S , Jariwala N , Wu A , Gandhi V et al . SMAD4 suppresses WNT-driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res 2018;78:4878–90.
- Shih IM , Wang TL , Traverso G , Romans K , Hamilton SR , Ben-Sasson S et al . Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A 2001;98:2640–5.
- Ordóñez-Morán P , Dafflon C , Imajo M , Nishida E , Huelsken J . HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 2015;28:815–29.
- Tan SH , Barker N . Stemming colorectal cancer growth and metastasis: HOXA5 forces cancer stem cells to differentiate. Cancer Cell 2015;28:683–5.
- Köhler C , Nittner D , Rambow F , Radaelli E , Stanchi F , Vandamme N et al . Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 2017;21:679–93.
- Shah M , Bhoumik A , Goel V , Dewing A , Breitwieser W , Kluger H et al . A role for ATF2 in regulating MITF and melanoma development. PLoS Genet 2010;6:e1001258.
- Claps G , Cheli Y , Zhang T , Scortegagna M , Lau E , Kim H et al . A transcriptionally inactive ATF2 variant drives melanomagenesis. Cell Rep 2016;15:1884–92.
- Saghafinia S , Homicsko K , Di Domenico A , Wullschleger S , Perren A , Marinoni I et al . Cancer cells retrace a stepwise differentiation program during malignant progression. Cancer Discov 2021;11:2638–57.
- Yu X-X , Qiu W-L , Yang L , Zhang Y , He M-Y , Li L-C et al . Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J 2019;38:e100164.
- de Thé H . Differentiation therapy revisited. Nat Rev Cancer 2018;18:117–27.
- He LZ , Merghoub T , Pandolfi PP . In vivo analysis of the molecular pathogenesis of acute promyelocytic leukemia in the mouse and its therapeutic implications. Oncogene 1999;18:5278–92.
- Warrell RP , de Thé H , Wang ZY , Degos L . Acute promyelocytic leukemia. N Engl J Med 1993;329:177–89.
- Bots M , Verbrugge I , Martin BP , Salmon JM , Ghisi M , Baker A et al . Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014;123:1341–52.
- Ferrara FF , Fazi F , Bianchini A , Padula F , Gelmetti V , Minucci S et al . Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res 2001;61:2–7.
- Kaufman CK , Mosimann C , Fan ZP , Yang S , Thomas AJ , Ablain J et al . A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016;351:aad2197.
- Morris JP , Yashinskie JJ , Koche R , Chandwani R , Tian S , Chen C-C et al . α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019;573:595–9.
- Saha SK , Parachoniak CA , Ghanta KS , Fitamant J , Ross KN , Najem MS et al . Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110–4.
- Dang L , Su S-SM . Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem 2017;86:305–31.
- Waitkus MS , Diplas BH , Yan H . Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018;34:186–95.
- Phan TG , Croucher PI . The dormant cancer cell life cycle. Nat Rev Cancer 2020;20:398–411.
- Jiang M , Azevedo-Pouly AC , Deering TG , Hoang CQ , DiRenzo D , Hess DA et al . MIST1 and PTF1 collaborate in feed-forward regulatory loops that maintain the pancreatic acinar phenotype in adult mice. Mol Cell Biol 2016;36:2945–55.
- Krah NM , Narayanan SM , Yugawa DE , Straley JA , Wright CVE , MacDonald RJ et al . Prevention and reversion of pancreatic tumorigenesis through a differentiation-based mechanism. Dev Cell 2019;50:744–54.
- Krah NM , De La O J-P , Swift GH , Hoang CQ , Willet SG , Chen Pan F et al . The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015;4:e07125.
- Shi G , DiRenzo D , Qu C , Barney D , Miley D , Konieczny SF . Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 2013;32:1950–8.
- Kopp JL , von Figura G , Mayes E , Liu F-F , Dubois CL , Morris JP et al . Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50.
- Julian LM , McDonald AC , Stanford WL . Direct reprogramming with SOX factors: masters of cell fate. Curr Opin Genet Dev 2017;46:24–36.
- Grimm D , Bauer J , Wise P , Krüger M , Simonsen U , Wehland M et al . The role of SOX family members in solid tumours and metastasis. Semin Cancer Biol 2020;67:122–53.
- Mu P , Zhang Z , Benelli M , Karthaus WR , Hoover E , Chen C-C et al . SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84–8.
- Von Hoff DD , LoRusso PM , Rudin CM , Reddy JC , Yauch RL , Tibes R et al . Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72.
- Biehs B , Dijkgraaf GJP , Piskol R , Alicke B , Boumahdi S , Peale F et al . A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018;562:429–33.
- Boumahdi S , de Sauvage FJ . The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19:39–56.
- Groves SM , Ireland A , Liu Q , Simmons AJ , Lau K , Iams WT et al . Cancer Hallmarks Define a Continuum of Plastic Cell States between Small Cell Lung Cancer Archetypes [Internet]. Systems Biology; 2021 Jan. Available from: http://biorxiv.org/lookup/doi/10.1101/2021.01.22.427865.
- LaFave LM , Kartha VK , Ma S , Meli K , Del Priore I , Lareau C et al . Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 2020;38:212–28.
- Marjanovic ND , Hofree M , Chan JE , Canner D , Wu K , Trakala M et al . Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 2020;38:229–46.
- Drapkin BJ , Minna JD . Studying lineage plasticity one cell at a time. Cancer Cell 2020;38:150–2.
- Inoue Y , Nikolic A , Farnsworth D , Liu A , Ladanyi M , Somwar R et al . Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer [Internet]. Cancer Biology; 2020 Nov. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.11.12.368522.
- Dravis C , Chung C-Y , Lytle NK , Herrera-Valdez J , Luna G , Trejo CL et al . Epigenetic and transcriptomic profiling of mammary gland development and tumor models disclose regulators of cell state plasticity. Cancer Cell 2018;34:466–82.
- Malta TM , Sokolov A , Gentles AJ , Burzykowski T , Poisson L , Weinstein JN et al . Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–54.
- Miao Z-F , Lewis MA , Cho CJ , Adkins-Threats M , Park D , Brown JW et al . A dedicated evolutionarily conserved molecular network licenses differentiated cells to return to the cell cycle. Dev Cell 2020;55:178–94.
- De Blander H , Morel A-P , Senaratne AP , Ouzounova M , Puisieux A . Cellular plasticity: a route to senescence exit and tumorigenesis. Cancers 2021;13:4561.
- Merrell AJ , Stanger BZ . Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 2016;17:413–25.
- Baylin SB , Jones PA . Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 2016;8:a019505.
- Flavahan WA , Gaskell E , Bernstein BE . Epigenetic plasticity and the hallmarks of cancer. Science 2017;357:eaal2380.
- Jones PA , Issa J-PJ , Baylin S . Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630–41.
- Huang S . Tumor progression: Chance and necessity in Darwinian and Lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol 2012;110:69–86.
- Darwiche N . Epigenetic mechanisms and the hallmarks of cancer: an intimate affair. Am J Cancer Res 2020;10:1954–78.
- Feng Y , Liu X , Pauklin S . 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell 2021;12:440–54.
- Nam AS , Chaligne R , Landau DA . Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet 2021;22:3–18.
- Bitman-Lotan E , Orian A . Nuclear organization and regulation of the differentiated state. Cell Mol Life Sci CMLS 2021;78:3141–58.
- Goldberg AD , Allis CD , Bernstein E . Epigenetics: a landscape takes shape. Cell 2007;128:635–8.
- Zeng Y , Chen T . DNA methylation reprogramming during mammalian development. Genes 2019;10:257.
- Hegde AN , Smith SG . Recent developments in transcriptional and translational regulation underlying long-term synaptic plasticity and memory. Learn Mem 2019;26:307–17.
- Kim S , Kaang B-K . Epigenetic regulation and chromatin remodeling in learning and memory. Exp Mol Med 2017;49:e281.
- Thienpont B , Van Dyck L , Lambrechts D . Tumors smother their epigenome. Mol Cell Oncol 2016;3:e1240549.
- Gameiro PA , Struhl K . Nutrient deprivation elicits a transcriptional and translational inflammatory response coupled to decreased protein synthesis. Cell Rep 2018;24:1415–24.
- Lin GL , Monje M . Understanding the deadly silence of posterior fossa A ependymoma. Mol Cell 2020;78:999–1001.
- Michealraj KA , Kumar SA , Kim LJY , Cavalli FMG , Przelicki D , Wojcik JB et al . Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell 2020;181:1329–45.
- Bakir B , Chiarella AM , Pitarresi JR , Rustgi AK . EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol 2020;30:764–76.
- Gupta PB , Pastushenko I , Skibinski A , Blanpain C , Kuperwasser C . Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 2019;24:65–78.
- Lambert AW , Weinberg RA . Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer 2021;21:325–38.
- Lindner P , Paul S , Eckstein M , Hampel C , Muenzner JK , Erlenbach-Wuensch K et al . EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis 2020;11:147.
- Javaid S , Zhang J , Anderssen E , Black JC , Wittner BS , Tajima K et al . Dynamic chromatin modification sustains epithelial-mesenchymal transition following inducible expression of Snail-1. Cell Rep 2013;5:1679–89.
- Serrano-Gomez SJ , Maziveyi M , Alahari SK . Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 2016;15:18.
- Skrypek N , Goossens S , De Smedt E , Vandamme N , Berx G . Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet TIG 2017;33:943–59.
- Li L , Hanahan D . Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 2013;153:86–100.
- Li L , Zeng Q , Bhutkar A , Galván JA , Karamitopoulou E , Noordermeer D et al . GKAP acts as a genetic modulator of NMDAR signaling to govern invasive tumor growth. Cancer Cell 2018;33:736–51.
- Mohammadi H , Sahai E . Mechanisms and impact of altered tumour mechanics. Nat Cell Biol 2018;20:766–74.
- Odenthal J , Takes R , Friedl P . Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis 2016;37:1117–28.
- Torres CM , Biran A , Burney MJ , Patel H , Henser-Brownhill T , Cohen A-HS et al . The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 2016;353:aaf1644.
- Puram SV , Tirosh I , Parikh AS , Patel AP , Yizhak K , Gillespie S et al . Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171:1611–24.
- Kinker GS , Greenwald AC , Tal R , Orlova Z , Cuoco MS , McFarland JM et al . Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 2020;52:1208–18.
- Murtha M , Esteller M . Extraordinary cancer epigenomics: thinking outside the classical coding and promoter box. Trends Cancer 2016;2:572–84.
- Nebbioso A , Tambaro FP , Dell’Aversana C , Altucci L . Cancer epigenetics: moving forward. PLoS Genet 2018;14:e1007362.
- Tavernari D , Battistello E , Dheilly E , Petruzzella AS , Mina M , Sordet-Dessimoz J et al . Non-genetic evolution drives lung adenocarcinoma spatial heterogeneity and progression. Cancer Discov 2021;11:1490–507.
- Heyn H , Vidal E , Ferreira HJ , Vizoso M , Sayols S , Gomez A et al . Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 2016;17:11.
- Saghafinia S , Mina M , Riggi N , Hanahan D , Ciriello G . Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 2018;25:1066–80.
- Audia JE , Campbell RM . Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8:a019521.
- Corces MR , Granja JM , Shams S , Louie BH , Seoane JA , Zhou W et al . The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898.
- Esteve-Puig R , Bueno-Costa A , Esteller M . Writers, readers and erasers of RNA modifications in cancer. Cancer Lett 2020;474:127–37.
- Janin M , Coll-SanMartin L , Esteller M . Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer 2020;19:70.
- Hanahan D , Coussens LM . Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–22.
- Lu Z , Zou J , Li S , Topper MJ , Tao Y , Zhang H et al . Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020;579:284–90.
- Thomas S , Izard J , Walsh E , Batich K , Chongsathidkiet P , Clarke G et al . The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res 2017;77:1783–812.
- Dzutsev A , Badger JH , Perez-Chanona E , Roy S , Salcedo R , Smith CK et al . Microbes and cancer. Annu Rev Immunol 2017;35:199–228.
- Helmink BA , Khan MAW , Hermann A , Gopalakrishnan V , Wargo JA . The microbiome, cancer, and cancer therapy. Nat Med 2019;25:377–88.
- Sears CL , Garrett WS . Microbes, microbiota, and colon cancer. Cell Host Microbe 2014;15:317–28.
- Pleguezuelos-Manzano C , Puschhof J , Rosendahl Huber A , van Hoeck A , Wood HM , Nomburg J et al . Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020;580:269–73.
- Okumura S , Konishi Y , Narukawa M , Sugiura Y , Yoshimoto S , Arai Y et al . Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat Commun 2021;12:5674.
- Salvi PS , Cowles RA . Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in homeostasis and disease. Cells 2021;10:1775.
- Fessler J , Matson V , Gajewski TF . Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 2019;7:108.
- Gopalakrishnan V , Helmink BA , Spencer CN , Reuben A , Wargo JA . The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 2018;33:570–80.
- Zitvogel L , Ma Y , Raoult D , Kroemer G , Gajewski TF . The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–70.
- Baruch EN , Youngster I , Ben-Betzalel G , Ortenberg R , Lahat A , Katz L et al . Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021;371:602–9.
- Davar D , Dzutsev AK , McCulloch JA , Rodrigues RR , Chauvin J-M , Morrison RM et al . Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021;371:595–602.
- Griffin ME , Espinosa J , Becker JL , Luo J-D , Carroll TS , Jha JK et al . Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 2021;373:1040–6.
- Mager LF , Burkhard R , Pett N , Cooke NCA , Brown K , Ramay H et al . Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369:1481–9.
- Ansaldo E , Belkaid Y . How microbiota improve immunotherapy. Science 2021;373:966–7.
- Ansaldo E , Farley TK , Belkaid Y . Control of immunity by the microbiota. Annu Rev Immunol 2021;39:449–79.
- Zhang Q , Ma C , Duan Y , Heinrich B , Rosato U , Diggs LP et al . Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov 2021;11:1248–67.
- Ding T , Schloss PD . Dynamics and associations of microbial community types across the human body. Nature 2014;509:357–60.
- Byrd AL , Liu M , Fujimura KE , Lyalina S , Nagarkar DR , Charbit B et al . Gut microbiome stability and dynamics in healthy donors and patients with non-gastrointestinal cancers. J Exp Med 2021;218:e20200606.
- Healy CM , Moran GP . The microbiome and oral cancer: more questions than answers. Oral Oncol 2019;89:30-3.
- Swaney MH , Kalan LR . Living in your skin: microbes, molecules and mechanisms. Infect Immun 2021;89:e00695–20.
- Willis JR , Gabaldón T . The human oral microbiome in health and disease: from sequences to ecosystems. Microorganisms 2020;8:308.
- Xu J , Peng J-J , Yang W , Fu K , Zhang Y . Vaginal microbiomes and ovarian cancer: a review. Am J Cancer Res 2020;10:743–56.
- Nejman D , Livyatan I , Fuks G , Gavert N , Zwang Y , Geller LT et al . The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80.
- Jin C , Lagoudas GK , Zhao C , Bullman S , Bhutkar A , Hu B et al . Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019;176:998–1013.
- Pushalkar S , Hundeyin M , Daley D , Zambirinis CP , Kurz E , Mishra A et al . The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 2018;8:403–16.
- McAllister F , Khan MAW , Helmink B , Wargo JA . The tumor microbiome in pancreatic cancer: bacteria and beyond. Cancer Cell 2019;36:577–9.
- Kadosh E , Snir-Alkalay I , Venkatachalam A , May S , Lasry A , Elyada E et al . The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020;586:133–8.
- Birch J , Gil J . Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34:1565–76.
- Faget DV , Ren Q , Stewart SA . Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 2019;19:439–53.
- Gorgoulis V , Adams PD , Alimonti A , Bennett DC , Bischof O , Bishop C et al . Cellular senescence: defining a path forward. Cell 2019;179:813–27.
- He S , Sharpless NE . Senescence in health and disease. Cell 2017;169:1000–11.
- Kowald A , Passos JF , Kirkwood TBL . On the evolution of cellular senescence. Aging Cell 2020;19:e13270.
- Lee S , Schmitt CA . The dynamic nature of senescence in cancer. Nat Cell Biol 2019;21:94–101.
- Wang B , Kohli J , Demaria M . Senescent cells in cancer therapy: friends or foes? Trends Cancer 2020;6:838–57.
- Baker DJ , Childs BG , Durik M , Wijers ME , Sieben CJ , Zhong J et al . Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–9.
- Ruhland MK , Loza AJ , Capietto A-H , Luo X , Knolhoff BL , Flanagan KC et al . Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 2016;7:11762.
- Hwang HJ , Lee Y-R , Kang D , Lee HC , Seo HR , Ryu J-K et al . Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett 2020;490:100–10.
- Wang D , Xiao F , Feng Z , Li M , Kong L , Huang L et al . Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res 2020;22:103.
- Amor C , Feucht J , Leibold J , Ho Y-J , Zhu C , Alonso-Curbelo D et al . Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020;583:127–32.
- Aster JC , Pear WS , Blacklow SC . The varied roles of notch in cancer. Annu Rev Pathol 2017;12:245–75.
- Kuczynski EA , Vermeulen PB , Pezzella F , Kerbel RS , Reynolds AR . Vessel co-option in cancer. Nat Rev Clin Oncol 2019;16:469–93.
Google ScholarCrossref