 Suche
Suche
 Mein Konto
Mein Konto
Caratteristiche del Cancro: nuove dimensioni

Prefazione Le caratteristiche distintive del cancro La concettualizzazione è uno strumento euristico per distillare l'enorme complessità dei fenotipi e dei genotipi del cancro in un insieme preliminare di principi sottostanti. Con l’avanzare della conoscenza dei meccanismi del cancro, altri aspetti della malattia sono emersi come potenziali miglioramenti. Ciò solleva la prospettiva che la plasticità fenotipica e la differenziazione disordinata siano un’abilità caratteristica distinta e che la riprogrammazione epigenetica non mutazionale e i microbiomi polimorfici rappresentino entrambi proprietà abilitanti caratteristiche che facilitano l’acquisizione di abilità caratteristiche. Inoltre, cellule senescenti di diversa origine possono essere aggiunte all'elenco dei tipi cellulari funzionalmente importanti nel microambiente tumorale. Significato Il cancro fa paura in...

Caratteristiche del Cancro: nuove dimensioni
Prefazione
La concettualizzazione degli elementi distintivi del cancro è uno strumento euristico per distillare l'enorme complessità dei fenotipi e dei genotipi del cancro in un insieme preliminare di principi sottostanti. Con l’avanzare della conoscenza dei meccanismi del cancro, altri aspetti della malattia sono emersi come potenziali miglioramenti. Ciò solleva la prospettiva che la plasticità fenotipica e la differenziazione disordinata siano un’abilità caratteristica distinta e che la riprogrammazione epigenetica non mutazionale e i microbiomi polimorfici rappresentino entrambi proprietà abilitanti caratteristiche che facilitano l’acquisizione di abilità caratteristiche. Inoltre, cellule senescenti di diversa origine possono essere aggiunte all'elenco dei tipi cellulari funzionalmente importanti nel microambiente tumorale.
Senso
Il cancro è spaventoso per l’ampiezza e la portata della sua diversità, che comprende la genetica, la biologia cellulare e tissutale, la patologia e la risposta alla terapia. Strumenti e tecnologie sperimentali e computazionali sempre più potenti stanno fornendo una valanga di “big data” sulla miriade di manifestazioni patologiche che il cancro comprende. Il concetto integrativo incorporato nei tratti distintivi del cancro aiuta a distillare questa complessità in una scienza sempre più logica, e le nuove dimensioni preliminari presentate in questa prospettiva possono aggiungere valore a questo sforzo per comprendere meglio i meccanismi della carcinogenesi e della progressione maligna e applicare questa conoscenza alla medicina del cancro.
introduzione
I segni distintivi del cancro sono stati proposti come un insieme di capacità funzionali che le cellule umane acquisiscono mentre passano dalla normalità agli stati di crescita neoplastica, più specificamente capacità critiche per la loro capacità di formare tumori maligni. In questi articoli ( 1, 2 ), Bob Weinberg e io abbiamo elencato ciò che immaginavamo come punti comuni che uniscono tutti i tipi di cellule tumorali a livello di fenotipo cellulare. L'intento era quello di fornire un quadro concettuale che consentisse di razionalizzare i complessi fenotipi di diversi tipi e varianti di tumore umano in relazione a un insieme comune di parametri cellulari sottostanti. Inizialmente, avevamo previsto l'inclusione complementare di sei diverse funzionalità del marchio e successivamente abbiamo ampliato questo numero a otto.
Questa formulazione è stata influenzata dal riconoscimento che i tumori umani si sviluppano come prodotti di processi multifase e che l’acquisizione di queste capacità funzionali potrebbe essere attribuita in qualche modo alle distinte fasi della patogenesi del tumore. La diversità della patogenesi maligna, che comprende molteplici tipi di tumore e una crescente pletora di sottotipi, comporta varie aberrazioni (e quindi abilità e proprietà acquisite) che sono il risultato di barriere tessuto-specifiche che vengono necessariamente aggirate durante alcuni percorsi di tumorigenesi. Sebbene riconosciamo che tali meccanismi specializzati possano essere utili, abbiamo limitato la designazione dei segni distintivi ai parametri che hanno un ampio impatto su tutto lo spettro dei tumori umani.
Gli otto tratti distintivi attualmente includono (Fig.1, a sinistra) le capacità acquisite di mantenere la segnalazione proliferativa, evitare i soppressori della crescita, resistere alla morte cellulare, consentire l'immortalità replicativa, indurre/accedere ai vasi, attivare l'invasione e le metastasi, riprogrammare il metabolismo cellulare ed evitare la distruzione del sistema immunitario. Nell’elaborazione più recente di questo concetto (2), la deregolamentazione del metabolismo cellulare e l’evitamento della distruzione del sistema immunitario sono stati definiti come “segni distintivi emergenti”, ma ora, undici anni dopo, è evidente che, analogamente ai sei originali, possono essere considerati segni distintivi fondamentali del cancro e sono inclusi come tali nella narrativa attuale (Fig. 1, a sinistra).
Figura 1
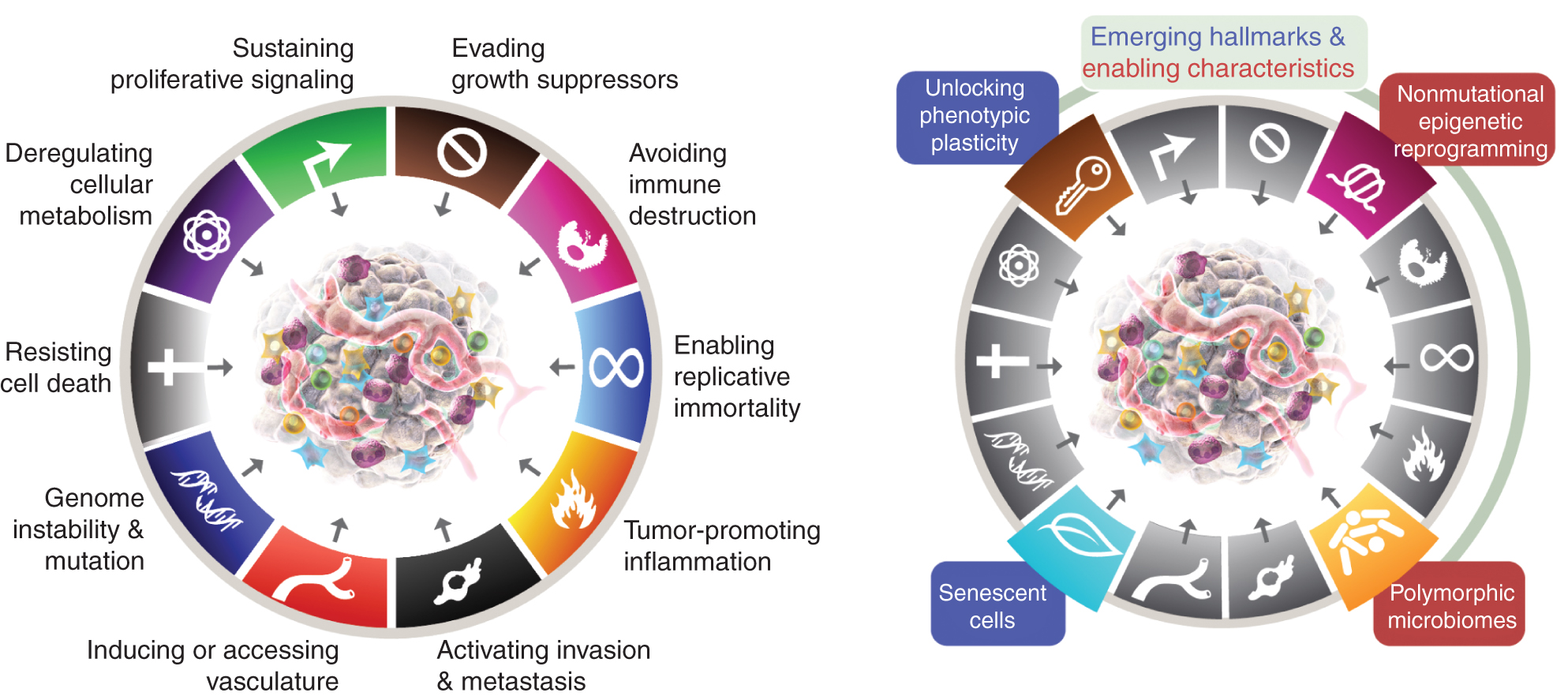
I tratti distintivi del Cancro attualmente incarnano otto abilità distintive e due qualità di supporto. Oltre alle sei capacità acquisite - caratteristiche distintive del cancro - proposte nel 2000 (1), le due "caratteristiche emergenti" preliminari introdotte nel 2011 (2) - l'energia cellulare (ora più comunemente chiamata "riprogrammazione del metabolismo cellulare") e "evitare la distruzione immunitaria" - sono state sufficientemente convalidate da essere considerate parte del nucleo fondamentale.
Dato il crescente riconoscimento che i tumori possono essere adeguatamente vascolarizzati, attivando l’angiogenesi o cooptando il sistema vascolare del tessuto normale (128), questo segno distintivo è anche definito in modo più ampio come la capacità di indurre o altrimenti accedere al sistema vascolare supportando la crescita del tumore principalmente attraverso l’invasione e la metastasi.
Il seguito del 2011 includeva anche “l’infiammazione che promuove il tumore” come secondo tratto abilitante, a complemento della generale “instabilità e mutazione del genoma”, che insieme erano fondamentalmente coinvolte nell’attivazione delle otto capacità (funzionali) necessarie per la crescita e la progressione del tumore. È vero, questa revisione include ulteriori nuovi tratti distintivi e caratteristiche abilitanti proposte, tra cui “sblocco della plasticità fenotipica”, “riprogrammazione epigenetica non mutazionale”, “microbiomi polimorfici” e “cellule senescenti”. I marchi della grafica del cancro sono stati adottati da Hanahan e Weinberg (2).
Come abbiamo notato all’epoca, queste caratteristiche distintive da sole non possono affrontare la complessità della patogenesi del cancro, vale a dire h. i precisi meccanismi molecolari e cellulari che consentono alle cellule preneoplastiche in via di sviluppo di sviluppare e acquisire queste capacità fenotipiche aberranti durante il corso della tumorigenesi e della progressione maligna.
Di conseguenza, abbiamo aggiunto un altro concetto alla discussione presentato come “caratteristiche abilitanti”, conseguenze dello stato aberrante della neoplasia che forniscono i mezzi attraverso i quali le cellule tumorali e i tumori possono acquisire queste caratteristiche funzionali. Pertanto, le proprietà abilitanti si riflettono nei meccanismi molecolari e cellulari attraverso i quali vengono acquisiti i tratti distintivi, piuttosto che nelle otto abilità stesse. Questi due processi di attivazione erano l’instabilità del genoma e l’infiammazione che promuove il tumore.
Abbiamo inoltre riconosciuto che il microambiente tumorale (TME), qui definito come composto da popolazioni eterogenee e interattive di cellule tumorali e cellule staminali tumorali insieme a una varietà di tipi di cellule stromali reclutate - il parenchima trasformato e lo stroma associato - è ora ampiamente apprezzato per svolgere un ruolo essenziale nella tumorigenesi e nella progressione maligna.
Dato il continuo interesse per queste formulazioni e la nostra continua intenzione di incoraggiare la discussione continua e il perfezionamento dello schema Hallmarks, è opportuno considerare una domanda frequente: ci sono ulteriori caratteristiche di questo modello concettuale che potrebbero essere incorporate, tenendo conto della necessità di garantirlo? che siano ampiamente applicabili in tutto lo spettro dei tumori umani? Di conseguenza, presenterò diversi potenziali nuovi tratti distintivi e caratteristiche abilitanti che potrebbero, a tempo debito, essere integrati come componenti fondamentali dei tratti distintivi della concettualizzazione del cancro.
Questi parametri sono "sblocco della plasticità fenotipica", "riprogrammazione epigenetica non mutazionale", "microbiomi polimorfici" e "cellule senescenti" (Fig. 1, a destra). È importante sottolineare che gli esempi presentati a sostegno di queste tesi sono illustrativi ma non esaustivi, poiché esiste un numero crescente e sempre più convincente di prove pubblicate a sostegno di ogni vignetta.
Sfruttare la plasticità fenotipica
Durante l'organogenesi, lo sviluppo, la determinazione e l'organizzazione delle cellule nei tessuti per svolgere funzioni omeostatiche è accompagnato dalla differenziazione terminale, con le cellule progenitrici che cessano di crescere, a volte in modo irreversibile, quando questi processi culminano. Pertanto, il risultato finale della differenziazione cellulare è, nella maggior parte dei casi, antiproliferativo, formando una chiara barriera alla continua proliferazione necessaria per la neoplasia.
Vi sono prove sempre più evidenti che sbloccare la capacità normalmente limitata della plasticità fenotipica di aggirare o sfuggire allo stato di differenziazione terminale è una componente critica della patogenesi del cancro (3). Questa plasticità può agire in diverse manifestazioni (Fig. 2). Pertanto, le cellule tumorali nascenti che hanno origine da una cellula normale che si è evoluta lungo un percorso che si avvicina o assume uno stato completamente differenziato possono invertire il corso dedifferenziandosi tornando a stati cellulari simili ai progenitori.
Al contrario, le cellule neoplastiche derivanti da una cellula progenitrice destinata a seguire un percorso che porta alla differenziazione terminale possono cortocircuitare il processo e mantenere le cellule tumorali in espansione in uno stato parzialmente differenziato, simile al progenitore. In alternativa, può verificarsi la transdifferenziazione in cui cellule originariamente impegnate in un percorso di differenziazione passano a un programma di sviluppo completamente diverso e quindi acquisiscono caratteristiche tessuto-specifiche che non erano predeterminate dalle loro normali cellule di origine.
I seguenti esempi supportano la tesi secondo cui diverse forme di plasticità cellulare rivelano plasticità fenotipica. A sinistra, la plasticità fenotipica è probabilmente un'abilità caratteristica acquisita che consente varie perturbazioni della differenziazione cellulare, tra cui (i) dedifferenziazione dagli stati maturi a quelli progenitori, (ii) differenziazione bloccata (terminale) dagli stati delle cellule progenitrici e (iii) transdifferenziazione in altri lignaggi cellulari. Sulla destra sono mostrate tre importanti modalità di differenziazione compromessa che sono parte integrante della patogenesi del cancro.
Pervertendo in modo differenziale la normale differenziazione delle cellule progenitrici in cellule mature nelle linee di sviluppo, vengono facilitate la tumorigenesi e la progressione maligna derivanti dalle cellule di origine in tali percorsi. I marchi della grafica del cancro sono stati adottati da Hanahan e Weinberg (2).
Figura 2

Dedifferenziazione
La carcinogenesi del colon è un esempio di differenziazione compromessa, poiché esiste la necessità teleologica che le cellule tumorali incipienti sfuggano al nastro trasportatore della differenziazione terminale e dell'esfoliazione, che potrebbe in linea di principio verificarsi attraverso la dedifferenziazione delle cellule epiteliali del colon che non si sono ancora differenziate terminalmente o attraverso la differenziazione bloccata delle cellule progenitrici/staminali nelle cripte che danno origine a queste cellule in differenziazione. Sia le cellule differenziate che le cellule staminali sono state implicate come cellule d'origine per il cancro del colon (4 – 6).
Due fattori di trascrizione dello sviluppo (TF), la proteina homeobox HOXA5 e SMAD4, quest'ultima coinvolta nella segnalazione BMP, sono altamente espressi nella differenziazione delle cellule epiteliali del colon e sono tipicamente persi nei carcinomi del colon avanzati, che tipicamente esprimono marcatori di cellule staminali e progenitrici. Perturbazioni funzionali nei modelli murini hanno dimostrato che l'espressione forzata di HOXA5 nelle cellule di cancro del colon ripristina i marcatori di differenziazione, sopprime i fenotipi delle cellule staminali e compromette l'invasione e le metastasi, fornendo una motivazione per la sua caratteristica downregulation (7, 8).
Al contrario, SMAD4 impone sia la differenziazione che la soppressione della proliferazione guidata dalla segnalazione oncogenica WNT, che è rivelata dalla perdita ingegnerizzata dell'espressione di SMAD4, fornendo una spiegazione per la sua perdita di espressione per consentire la dedifferenziazione e successivamente l'iperproliferazione guidata da WNT (5).
In particolare, la perdita di questi due “soppressori della differenziazione” con la conseguente dedifferenziazione è associata all’acquisizione di altre capacità di segno distintivo, così come di altri regolatori che inducono un segno distintivo, complicando la rigorosa definizione di questo segno distintivo provvisorio come separabile e indipendente.
Un'altra linea di evidenza riguarda la soppressa espressione del MITF, il regolatore principale della differenziazione dei melanociti, che sembra essere coinvolto nella genesi delle forme aggressive di melanoma maligno. La perdita di questo TF evolutivo è associata alla riattivazione dei geni progenitori della cresta neurale e alla downregulation dei geni che caratterizzano i melanociti completamente differenziati. La ricomparsa dei geni della cresta neurale indica che queste cellule ritornano allo stato progenitore da cui derivano i melanociti durante lo sviluppo.
Inoltre, uno studio di tracciamento del lignaggio dei melanomi indotti da BRAF ha stabilito che i melanociti pigmentati maturi sono cellule di origine che vanno incontro a dedifferenziazione durante il corso della tumorigenesi (9). In particolare, l’oncogene mutante BRAF, presente in più della metà dei melanomi cutanei, induce iperproliferazione, che precede e può quindi essere meccanicamente separata dalla successiva dedifferenziazione che deriva dalla downregulation del MITF.
Un altro studio ha implicato funzionalmente la sovraregolazione del TF ATF2 dello sviluppo, la cui espressione caratteristica nei melanomi murini e umani sopprime indirettamente MITF1, in concomitanza con la progressione maligna delle cellule di melanoma conseguentemente indifferenziate (10). Al contrario, l'espressione nei melanomi di forme mutanti di ATF2 che non possono sopprimere il MITF si traduce in melanomi ben differenziati (11).
Inoltre, uno studio recente (12) ha collegato la dedifferenziazione del lignaggio alla progressione maligna delle neoplasie delle cellule delle isole pancreatiche ai carcinomi predisposti alle metastasi; queste cellule neuroendocrine e i tumori derivati derivano da un lignaggio evolutivo distinto da quello che genera il numero molto maggiore di cellule vicine che formano l'esocrino e il pancreas e i conseguenti adenocarcinomi duttali.
Sorprendentemente, il percorso di differenziazione in più fasi dalle cellule progenitrici delle isole alle cellule β mature è stato completamente caratterizzato (13). Il profilo comparativo del trascrittoma mostra che i tumori delle isole simili a adenoma sono più simili alle cellule β immature ma differenziate produttrici di insulina, mentre i carcinomi invasivi sono più simili ai precursori delle cellule delle isole embrionali. La progressione verso carcinomi scarsamente differenziati comporta una fase iniziale di dedifferenziazione, che inizialmente non comporta un aumento della proliferazione o una ridotta apoptosi rispetto agli adenomi ben differenziati, che tendono entrambi a verificarsi successivamente.
Pertanto, il passaggio discreto della dedifferenziazione non è guidato da cambiamenti osservabili nelle caratteristiche caratteristiche della proliferazione sostenuta e della resistenza all’apoptosi. Piuttosto, la sovraregolazione di un miRNA precedentemente implicato nella specificazione dello stato progenitore delle isole è quella che viene sottoregolata durante la differenziazione terminale delle cellule β, 12).
Differenziazione bloccata
Mentre gli esempi sopra riportati illustrano come la soppressione dell'espressione del fattore di differenziazione può facilitare la tumorigenesi consentendo a cellule meglio differenziate di dedifferenziarsi in progenitori, in altri casi cellule progenitrici non completamente differenziate possono subire cambiamenti regolatori che bloccano attivamente la loro ulteriore progressione verso stati completamente differenziati, tipicamente non proliferativi.
È stato a lungo documentato che la leucemia promielocitica acuta (APL) deriva da una traslocazione cromosomica che fonde il locus PML con il gene che codifica per il recettore nucleare α dell'acido retinoico (RARα). Le cellule progenitrici mieloidi che trasportano tali traslocazioni sono apparentemente incapaci di continuare la loro consueta differenziazione terminale in granulociti, con il risultato che le cellule sono intrappolate in uno stadio progenitore proliferativo, simile ai promielociti ( 14 ).
La prova di questo schema deriva dal trattamento di cellule APL in coltura, modelli murini della malattia e pazienti affetti con acido retinoico, il ligando di RARα; Questo trattamento terapeutico induce le cellule neoplastiche dell'APL a differenziarsi in granulociti apparentemente maturi e non proliferanti, cortocircuitando così la loro progressiva espansione proliferativa (14-16).
Una variazione su questo tema riguarda un'altra forma di leucemia mieloide acuta, questa portatrice della traslocazione t(8;21) che produce la proteina di fusione AML1-ETO. Questa proteina da sola può trasformare i progenitori mieloidi, almeno in parte bloccandone la differenziazione. L'intervento terapeutico in modelli murini e pazienti con un inibitore farmacologico di una deacetilasi istonica che modifica la cromatina (HDAC) fa sì che le cellule di leucemia mieloide riprendano la differenziazione in cellule con una morfologia cellulare mieloide più matura. Ad accompagnare questa reazione c'è una riduzione della capacità proliferativa, compromettendo così la progressione di questa leucemia (17, 18).
Un terzo esempio nel melanoma riguarda un TF dello sviluppo, SOX10, che normalmente viene sottoregolato durante la differenziazione dei melanociti. Studi sul guadagno e sulla perdita di funzione in un modello di pesce zebra di melanomi indotti da BRAF hanno dimostrato che l'espressione anormalmente mantenuta di SOX10 blocca la differenziazione delle cellule progenitrici neurali in melanociti, consentendo la formazione di melanomi guidati da BRAF (19).
Altri esempi di modulatori della differenziazione includono il metabolita alfa-chetoglutarato (αKG), un cofattore necessario per un numero di enzimi che modificano la cromatina che ha dimostrato di essere coinvolto nella stimolazione di alcuni stati cellulari differenziati. Nel cancro del pancreas, il soppressore tumorale p53 stimola la produzione di αKG e il mantenimento di uno stato cellulare più differenziato, mentre una perdita prototipica della funzione p53 porta ad una riduzione dei livelli di αKG e alla conseguente dedifferenziazione, che è associata alla progressione maligna (20).
In una forma di cancro al fegato, la mutazione di un gene dell'isocitrato deidrogenasi (IDH1/2) non determina la produzione di αKG che induce la differenziazione, ma piuttosto un "oncometabolita" correlato, D-2-idrossigluterato (D2HG), che ha dimostrato di bloccare la differenziazione degli epatociti delle cellule progenitrici del fegato attraverso la repressione mediata da D2HG di un regolatore principale della differenziazione e della quiescenza degli epatociti, HNF4a.
La soppressione mediata da D2HG della funzione HNF4a innesca un'espansione proliferativa delle cellule progenitrici degli epatociti nel fegato, che diventano suscettibili alla trasformazione oncogenica dopo la successiva attivazione mutazionale dell'oncogene KRAS, che guida la progressione maligna verso il colangiocarcinoma del fegato (21). Il mutante IDH1/2 e il suo oncometabolita D2HG funzionano anche in una varietà di tumori mieloidi e altri tipi solidi, dove D2HG inibisce le diossigenasi αKG-dipendenti necessarie per gli eventi di metilazione dell'istone e del DNA che mediano i cambiamenti nella struttura della cromatina durante la differenziazione del lignaggio dello sviluppo, congelando così l'insorgenza delle cellule tumorali in uno stato progenitore (22, 23).
Un ulteriore concetto correlato è la “differenziazione bypassata”, in cui cellule progenitrici/staminali parzialmente o indifferenziate escono dal ciclo cellulare e giacciono dormienti in nicchie protettive, con il potenziale di riavviare l’espansione proliferativa (24), sebbene con ancora la pressione selettiva per interrompere la loro differenziazione programmata in un modo o nell’altro.
Transdifferenziazione
Il concetto di transdifferenziazione è stato riconosciuto da tempo dai patologi sotto forma di metaplasia tissutale, in cui cellule di un particolare fenotipo differenziato cambiano marcatamente la loro morfologia per diventare chiaramente riconoscibili come elementi di un altro tessuto, un esempio importante del quale è l'esofago di Barrett, dove l'infiammazione cronica dell'epitelio squamoso stratificato dell'esofago induce la transdifferenziazione in un epitelio colonnare semplice caratteristico dell'intestino, facilitando così il successivo sviluppo di adenocarcinomi piuttosto che carcinomi a cellule squamose attesi da questo epitelio squamoso (3).
Ora, i determinanti molecolari stanno rivelando meccanismi di transdifferenziazione in vari tumori, sia per i casi in cui la metaplasia tissutale grossolana è evidente, sia per altri in cui è un po’ più subdola, come illustrano i seguenti esempi.
Un caso informativo per la transdifferenziazione come evento discreto nella tumorigenesi riguarda l'adenocarcinoma duttale pancreatico (PDAC), in cui una delle cellule di origine coinvolte, la cellula acinosa pancreatica, può transdifferenziarsi in un fenotipo di cellula duttale durante l'inizio dello sviluppo neoplastico. Due TF, PTF1a e MIST1, controllano la specificazione e il mantenimento dello stato differenziato delle cellule acinose del pancreas attraverso la loro espressione nel contesto di circuiti regolatori “feed-forward” autosufficienti (25).
Entrambi questi TF sono spesso sottoregolati durante lo sviluppo neoplastico e la progressione maligna del PDAC umano e murino. Studi genetici funzionali su topi e cellule PDAC umane in coltura hanno dimostrato che l'espressione forzata sperimentalmente di PTF1a compromette la transdifferenziazione e la proliferazione indotte da KRAS e può anche forzare la ridifferenziazione di cellule già neoplastiche in un fenotipo di cellule acinose quiescenti (26).
Al contrario, la soppressione dell'espressione di PTF1a innesca la metaplasia da acino a dotto, vale a dire la transdifferenziazione, e quindi sensibilizza le cellule simili al dotto alla trasformazione oncogenica del KRAS, accelerando il successivo sviluppo del PDAC invasivo (27). Allo stesso modo, l'espressione forzata di MIST1 nel pancreas che esprime KRAS blocca anche la transdifferenziazione e compromette l'inizio della tumorigenesi pancreatica, che è altrimenti facilitata dalla formazione di lesioni precancerose simili a condotti (PanIN), mentre la delezione genetica di MIST1 migliora la loro formazione e l'inizio della progressione neoplastica guidata da KRAS (28).
La perdita dell'espressione di PTF1 o MIST1 durante la tumorigenesi è associata ad un aumento dell'espressione di un altro TF regolatore dello sviluppo, SOX9, che normalmente è efficace nella specificazione delle cellule duttali (27, 28). È stato anche dimostrato che la sovraregolazione forzata di SOX9, evitando così la necessità di downregolazione di PTF1a, e MIST1 stimola la transdifferenziazione delle cellule acinari in un fenotipo di cellule duttali sensibili alla neoplasia indotta da KRAS (29), implicando SOX9 come un effettore funzionale chiave della loro downregulation nella genesi del PDAC umano.
Pertanto, tre TF che regolano la differenziazione pancreatica possono essere alterati in vari modi per indurre uno stato transdifferenziato che, nel contesto dell'attivazione mutazionale di KRAS, facilita la trasformazione oncogenica e l'inizio della tumorigenesi e della progressione maligna.
Ulteriori membri della famiglia SOX di fattori regolatori associati alla cromatina sono, da un lato, ampiamente associati sia alla specifica del destino cellulare che al cambio di lignaggio in fase di sviluppo ( 30 ) e, dall'altro, a diversi fenotipi associati al tumore ( 31 ). Un altro importante esempio di transdifferenziazione mediata da SOX coinvolge un meccanismo di resistenza terapeutica nel cancro alla prostata.
In questo caso, la perdita dei soppressori tumorali RB e p53 - la cui assenza è caratteristica dei tumori neuroendocrini - in risposta alla terapia antiandrogena è necessaria ma non sufficiente per la trasformazione comunemente osservata di cellule tumorali della prostata ben differenziate in cellule di carcinoma che hanno invaso il lignaggio di differenziazione con caratteristiche molecolari e istologiche delle cellule neuroendocrine che, in particolare, non esprimono il recettore degli androgeni. Oltre alla perdita di RB e p53, la resistenza acquisita alla terapia antiandrogena richiede un'espressione sovraregolata di SOX2, un gene regolatore dello sviluppo, che ha dimostrato di aiutare a indurre la transdifferenziazione delle cellule di adenocarcinoma sensibili alla terapia in derivati che si trovano in uno stato cellulare neuroendocrino refrattario alla terapia (32).
Un terzo esempio mostra anche la transdifferenziazione come strategia utilizzata dalle cellule di carcinoma per evitare l'eliminazione mediante terapia specifica per il lignaggio, in questo caso con carcinomi a cellule basali (BCC) della pelle trattati con un inibitore farmacologico della via oncogena Hedgehog-Smoothened (HH/SMO) nota per guidare la crescita neoplastica di queste cellule ( 33 ).
Le cellule tumorali resistenti ai farmaci passano a un tipo cellulare correlato allo sviluppo ma distinto attraverso ampi cambiamenti epigenetici in specifici domini della cromatina e un'alterata accessibilità di due superpotenziatori. Lo stato fenotipico appena acquisito delle cellule BCC consente loro di mantenere l'espressione della via di segnalazione oncogenica WNT, che a sua volta conferisce l'indipendenza dalla via di segnalazione HH/SMO soppressa dai farmaci (34).
Come previsto da questa transdifferenziazione, il trascrittoma delle cellule tumorali si sposta da una firma genetica che riflette la cellula coinvolta di origine dei BCC, vale a dire le cellule staminali del rigonfiamento del follicolo pilifero, a una firma indicativa delle cellule staminali basali che popolano l'epidermide interfollicolare del BCC. Tale transdifferenziazione per consentire la resistenza ai farmaci è sempre più documentata in varie forme di cancro (35).
La plasticità del lignaggio evolutivo sembra essere prevalente anche nei principali sottotipi di carcinoma polmonare, vale a dire h. nei carcinomi neuroendocrini [carcinoma polmonare a piccole cellule (SCLC)] e adenocarcinomi + carcinomi a cellule squamose [carcinoma polmonare collettivo non a piccole cellule (NSCLC)]. Il sequenziamento dell'RNA di singole cellule ha rivelato una conversione notevolmente dinamica ed eterogenea tra questi sottotipi, nonché marcate variazioni al loro interno, durante le fasi della tumorigenesi polmonare, la successiva progressione maligna e la risposta alla terapia ( 36 – 38 ).
Pertanto, piuttosto che la semplice concettualizzazione di un puro passaggio clonale da un lignaggio all’altro, questi studi dipingono un quadro molto più complesso di sottopopolazioni di cellule tumorali che si interconvertono dinamicamente che mostrano caratteristiche di molteplici lignaggi di sviluppo e stadi di differenziazione, una visione che fa riflettere a questo riguardo per il targeting terapeutico basato sul lignaggio del cancro del polmone umano. I determinanti regolatori di questa plasticità fenotipica dinamica stanno cominciando ad essere identificati (37, 39, 40).
Riepilogo
Le tre classi di meccanismi sopra descritti evidenziano regolatori selettivi della plasticità cellulare che sono – almeno parzialmente – separabili dai principali fattori oncogeni e da altre capacità distintive. Al di là di questi esempi, esiste un insieme significativo di prove che collegano molte forme di cancro a una differenziazione compromessa, che è accompagnata dall'acquisizione di firme trascrittomiche e altri fenotipi - ad esempio, morfologia istologica - associati agli stadi di cellule progenitrici o staminali osservati nei corrispondenti tessuti normali. origine o in altri tipi cellulari e lignaggi più lontani (41 – 43).
Pertanto, queste tre sottoclassi di plasticità fenotipica - dedifferenziazione delle cellule mature allo stato progenitore, differenziazione in stallo per congelare le cellule in via di sviluppo negli stati progenitore/staminale e transdifferenziazione verso lignaggi cellulari alternativi - sembrano essere efficaci in diversi tipi di cancro durante la tumorigenesi primaria, la progressione maligna e/o la risposta alla terapia.
Ci sono però due considerazioni concettuali. In primo luogo, la dedifferenziazione e la differenziazione bloccata sono probabilmente intrecciate, poiché sono indistinguibili in molti tipi di tumore in cui la cellula di origine – cellula differenziata o cellula progenitrice/staminale – è sconosciuta o alternativamente coinvolta. In secondo luogo, l’acquisizione o il mantenimento dei fenotipi delle cellule progenitrici e la perdita di caratteristiche differenziate è, nella maggior parte dei casi, un riflesso impreciso del normale stadio di sviluppo, immergendosi in un ambiente di altri cambiamenti caratteristici nella cellula tumorale che non sono presenti nelle cellule in via di sviluppo naturale.
Inoltre, un'altra forma di plasticità fenotipica coinvolge la senescenza cellulare, discussa più in generale di seguito, per cui le cellule tumorali indotte a subire un invecchiamento apparentemente irreversibile sono invece in grado di sfuggire e continuare l'espansione proliferativa (44). Infine, come per altre capacità distintive, la plasticità cellulare non è una nuova invenzione o un’aberrazione delle cellule tumorali, ma piuttosto la corruzione di capacità latenti ma attivabili che varie cellule normali utilizzano per supportare l’omeostasi, la riparazione e la rigenerazione (45).
Nel complesso, questi esempi illustrativi incoraggiano la considerazione che sbloccare la plasticità cellulare per consentire varie forme di differenziazione perturbata rappresenta una capacità distintiva distinta che differisce nella regolazione e nel fenotipo cellulare dai tratti distintivi fondamentali ben convalidati del cancro (Fig. 2).
Riprogrammazione epigenetica senza mutazione
La proprietà abilitante dell’instabilità e della mutazione del genoma (DNA) è una componente fondamentale dello sviluppo e della patogenesi del cancro. Attualmente, diversi consorzi internazionali stanno catalogando le mutazioni in tutto il genoma delle cellule tumorali umane, praticamente in ogni tipo di cancro umano, nei vari stadi della progressione maligna, comprese le lesioni metastatiche, e durante lo sviluppo della resistenza alla terapia adattativa. Un risultato è il riconoscimento ormai diffuso che le mutazioni nei geni che organizzano, modulano e mantengono l’architettura della cromatina e quindi regolano l’espressione genetica a livello globale vengono sempre più scoperte e funzionalmente collegate ai tratti del cancro (46 – 48).
Inoltre, ci sono argomenti a favore di un’altra forma apparentemente indipendente di riprogrammazione del genoma che coinvolge cambiamenti regolati puramente epigeneticamente nell’espressione genica, che potrebbe essere definita “riprogrammazione epigenetica non mutazionale” (Fig. 3). In effetti, la tesi dell’evoluzione del cancro senza mutazioni e della programmazione puramente epigenetica dei fenotipi caratteristici del cancro è stata sollevata quasi un decennio fa (49) ed è sempre più discussa (46, 50–52).
Figura 3

Similmente a quanto avviene durante l’embriogenesi, la differenziazione dei tessuti e l’omeostasi, le prove accumulate suggeriscono che i circuiti e le reti di regolazione genica strumentale nei tumori possono essere controllati da una pletora di meccanismi corrotti e cooptati che sono indipendenti dall’instabilità del genoma e dalla mutazione genetica. I marchi della grafica del cancro sono stati adottati da Hanahan e Weinberg (2).
Naturalmente, il concetto di regolazione epigenetica non mutazionale dell'espressione genica è ben consolidato come meccanismo centrale che media lo sviluppo embrionale, la differenziazione e l'organogenesi ( 53 – 55 ). Nell'adulto, ad esempio, la memoria a lungo termine comporta cambiamenti nella modificazione dei geni e degli istoni, nella struttura della cromatina e nell'attivazione degli interruttori di espressione genetica, che sono mantenuti stabilmente nel tempo da circuiti di feedback positivi e negativi (56, 57). Prove crescenti supportano l’idea che analoghi cambiamenti epigenetici possano contribuire all’acquisizione di abilità caratteristiche durante lo sviluppo del tumore e la progressione maligna. A sostegno di questa ipotesi si riportano di seguito alcuni esempi.
Meccanismi microambientali di riprogrammazione epigenetica
Se non solo attraverso mutazioni oncogene, come viene riprogrammato il genoma delle cellule tumorali? Un numero crescente di prove suggerisce che le proprietà fisiche aberranti del microambiente tumorale possono causare ampi cambiamenti nell’epigenoma, di cui alterazioni benefiche per la selezione fenotipica delle capacità dei tratti possono portare alla crescita clonale delle cellule tumorali con una migliore idoneità all’espansione proliferativa.
Una caratteristica comune dei tumori (o delle regioni all'interno dei tumori) è l'ipossia come risultato di una vascolarizzazione inadeguata. L'ipossia, ad esempio, riduce l'attività delle demetilasi TET, portando a cambiamenti significativi nel metiloma, in particolare all'ipermetilazione (58). È probabile anche che una vascolarizzazione insufficiente limiti la biodisponibilità dei nutrienti essenziali trasportati dal sangue e, ad esempio, è stato dimostrato che la privazione dei nutrienti altera il controllo traslazionale e di conseguenza aumenta il fenotipo maligno delle cellule del cancro al seno (59).
Un esempio convincente di regolazione epigenetica mediata dall’ipossia è una forma di ependimoma pediatrico invariabilmente fatale. Come molti tumori embrionali e pediatrici, questa forma è priva di mutazioni ricorrenti, in particolare della mancanza di mutazioni driver negli oncogeni e nei soppressori tumorali. Piuttosto, è stato dimostrato che la crescita anormale di queste cellule tumorali è controllata da un programma di regolazione genetica indotto dall’ipossia (60, 61). Sorprendentemente, la presunta cellula di origine di questo cancro risiede in un compartimento ipossico e probabilmente sensibilizza le cellule al suo interno per avviare la tumorigenesi attraverso cofattori ancora sconosciuti.
Un’altra prova convincente della regolazione epigenetica mediata dal microambiente riguarda la capacità di crescita invasiva delle cellule tumorali. Un classico esempio è l'induzione reversibile dell'invasività delle cellule tumorali ai margini di molti tumori solidi, orchestrata dal programma di regolamentazione dello sviluppo noto come transizione epiteliale-mesenchimale (EMT; rif. 62–64). In particolare, è stato recentemente dimostrato che un regolatore principale di EMT, ZEB1, induce l'espressione di un'istone metiltransferasi, SETD1B, che a sua volta mantiene l'espressione di ZEB1 in un circuito di feedback positivo che mantiene lo stato regolatorio (invasivo) di EMT (65).
Uno studio precedente ha documentato in modo simile che l'induzione di EMT attraverso l'espressione sovraregolata di un TF correlato, SNAIL1, ha causato marcati cambiamenti nel panorama della cromatina come risultato dell'induzione di un numero di modificatori della cromatina la cui attività si è dimostrata necessaria per il mantenimento dello stato fenotipico (66). Inoltre, una serie di condizioni e fattori sperimentati dalle cellule tumorali ai margini dei tumori, tra cui l’ipossia e le citochine secrete dalle cellule stromali, possono apparentemente indurre EMT e quindi invasività (67, 68).
Un esempio lampante di programmazione dell'invasività da parte del microambiente, presumibilmente non correlato al programma EMT, comporta l'attivazione autocrina di un circuito di segnalazione neuronale che coinvolge il glutammato secreto e il suo recettore NMDAR (69, 70). Sorprendentemente, la rigidità prototipica di molti tumori solidi, incarnata in estese alterazioni della matrice extracellulare (ECM) che racchiude le cellule al loro interno, ha profonde implicazioni per le proprietà invasive e altre proprietà fenotipiche delle cellule tumorali.
Rispetto all'ECM del tessuto normale da cui originano i tumori, l'ECM tumorale è tipicamente caratterizzata da maggiore reticolazione e densità, modifiche enzimatiche e composizione molecolare alterata che orchestrano collettivamente, in parte attraverso recettori integrinici per motivi ECM, reti di segnalazione e di espressione genica indotte dalla rigidità che inducono invasività e altre caratteristiche (71).
Oltre a tali meccanismi di regolazione forniti dal microambiente fisico del tumore, la segnalazione paracrina, comprendente fattori solubili rilasciati nell'ambiente extracellulare dai vari tipi di cellule che popolano i tumori solidi, può anche contribuire all'induzione di diversi programmi di crescita invasiva morfologicamente distinti (72), solo uno dei quali - chiamato "mesenchimale" - sembra essere coinvolto nel suddetto meccanismo di regolazione epigenetica dell'EMT.
Eterogeneità regolatoria epigenetica
Una crescente base di conoscenze aumenta l'apprezzamento per l'importanza dell'eterogeneità intratumorale nel generare la diversità fenotipica in cui le cellule più adatte per l'espansione proliferativa e l'invasione superano i loro fratelli e vengono quindi selezionate per la progressione maligna. Certamente, un aspetto di questa eterogeneità fenotipica è dovuto all’instabilità genomica cronica o episodica e alla conseguente eterogeneità genetica nelle cellule che popolano un tumore.
Inoltre, sta diventando sempre più chiaro che può esistere un'eterogeneità epigenetica non basata sulla mutazione. Un esempio importante è l’istone linker H1.0, che è espresso e represso dinamicamente in sottopopolazioni di cellule tumorali all’interno di una gamma di tipi di tumore, con conseguente sequestro o accessibilità di domini di dimensioni megabasi [73]. In particolare, è stato scoperto che la popolazione di cellule tumorali con H1.0 represso mostra proprietà simili a quelle staminali, una maggiore capacità di avviare il tumore e un'associazione con una prognosi sfavorevole nei pazienti.
Un altro esempio di plasticità regolata epigeneticamente è stato descritto nei carcinomi orali umani a cellule squamose (SCC), dove le cellule tumorali ai margini invasivi adottano uno stato EMT parziale (p-EMT) che manca dei suddetti TF mesenchimali ma esprime altri geni che definiscono EMT che non sono espressi nel nucleo centrale dei tumori (74).
Le cellule p-EMT ovviamente non rappresentano la compartimentazione clonale delle cellule alterate per mutazione: le colture di cellule tumorali primarie derivate dal tumore contengono miscele dinamiche di cellule p-EMT hi e p-EMT lo e quando le cellule p-EMT hi/lo sono state purificate e coltivate tramite FACS, entrambe sono ritornate a popolazioni miste di p-EMT hi e p-EMT lo entro 4 giorni. Sebbene i segnali paracrini provenienti dallo stroma adiacente possano essere considerati deterministici per lo stato p-EMT hi, la presenza stabile e la rigenerazione dei due stati epigenetici nella coltura sostengono un meccanismo intrinseco alle cellule tumorali. In particolare, questa conclusione è supportata dall'analisi di 198 linee cellulari che rappresentano 22 tipi di cancro, incluso l'SCC, dove 12 stati epigenetici stabilmente eterogenei (incluso il p-EMT nell'SCC) sono stati variamente rilevati nei modelli di linee cellulari così come i relativi tumori primari (75).
Ancora una volta, gli stati fenotipici eterogenei non possono essere collegati a differenze genetiche rilevabili, e in diversi casi è stato dimostrato che le cellule selezionate tramite FACS di un particolare stato si riequilibrano dinamicamente sulla coltura, ricapitolando un equilibrio stabile tra gli stati eterogenei osservati nelle linee cellulari originali.
Inoltre, le tecnologie per la profilazione dell’intero genoma di vari attributi – oltre la sequenza del DNA e la sua variazione mutazionale – mettono in luce elementi influenti dell’annotazione e dell’organizzazione del genoma delle cellule tumorali che sono correlati alla prognosi del paziente e, sempre più, alle abilità caratteristiche (76 – 78). L'eterogeneità epigenomica viene rivelata da tecnologie sempre più potenti per la profilazione della metilazione del DNA a livello genomico (79, 80), la modificazione degli istoni (81), l'accessibilità della cromatina (82) e la modifica post-trascrizionale e la traduzione dell'RNA (83, 84).
Una sfida rispetto al postulato qui considerato sarà quella di determinare quali modifiche epigenomiche in alcuni tipi di cancro (i) hanno un significato normativo e (ii) sono rappresentative di una riprogrammazione puramente non mutazionale, in contrapposizione all'instabilità guidata dalla mutazione e quindi spiegabile dal genoma.
Regolazione epigenetica dei tipi di cellule stromali che popolano il microambiente tumorale
In generale, non si ritiene che le cellule accessorie nel microambiente tumorale che contribuiscono funzionalmente all'acquisizione di abilità caratteristiche soffrano di instabilità genetica e riprogrammazione mutazionale per migliorare le loro attività di promozione del tumore; piuttosto, si è concluso che queste cellule – fibroblasti associati al cancro, cellule immunitarie innate, cellule endoteliali e periciti del sistema vascolare tumorale – sono riprogrammate epigeneticamente al momento del loro reclutamento da parte di fattori solubili e fisici che definiscono il microambiente tumorale solido (2, 85).
Si prevede che le tecnologie di profilazione multi-omica attualmente applicate alle cellule tumorali saranno sempre più utilizzate per studiare le cellule accessorie (stromali) nei tumori per chiarire come le cellule normali vengono danneggiate per supportare funzionalmente lo sviluppo e la progressione del tumore. Ad esempio, uno studio recente (86) suggerisce che tale riprogrammazione può comportare modifiche dell’epigenoma, oltre allo scambio induttivo di citochine, chemochine e fattori di crescita che alterano le reti di segnalazione intracellulare in tutti questi tipi di cellule:
Quando modelli murini con metastasi polmonari sono stati trattati con una combinazione di un inibitore della DNA metiltransferasi (5-azacitidina) e un inibitore della modificazione degli istoni (un HDAC), si è scoperto che le cellule mieloidi infiltranti erano passate da uno stato progenitore immaturo (promotore del tumore) a cellule somiglianti a macrofagi interstiziali maturi (antagonisti del tumore), che, a differenza delle loro controparti nei tumori non trattati, non erano in grado di supportare le tipiche capacità richieste per un efficiente colonizzazione metastatica ( 86). È concepibile che la profilazione multi-omica e le perturbazioni farmacologiche servano a chiarire lo stato epigenetico riprogrammato in tali cellule mieloidi così come in altri tipi cellulari accessori caratteristici che popolano i microambienti tumorali.
Riepilogo
Nel loro insieme, queste istantanee illustrative supportano la tesi secondo cui la riprogrammazione epigenetica senza mutazione sarà accettata come un vero tratto abilitante che serve a facilitare l'acquisizione di abilità caratteristiche (Fig. 3), distinte dall'instabilità e dalla mutazione del DNA genomico. In particolare, ci si può aspettare che la riprogrammazione epigenetica non mutazionale si dimostri fondamentale per abilitare la nuova capacità distintiva preliminare della plasticità fenotipica discussa sopra, in particolare come forza trainante nell'eterogeneità trascrittomica dinamica che è sempre più ben documentata nelle TME delle cellule tumorali maligne. Il progresso delle tecnologie di profilazione multi-omica unicellulare farà luce sui rispettivi contributi e sull'interazione tra la regolazione epigenetica guidata dalla mutazione e non guidata dalla mutazione nello sviluppo dei tumori durante la progressione maligna e la metastasi.
Microbiomi polimorfici
Una frontiera di vasta portata nella biomedicina si sta aprendo mettendo in luce la diversità e la variabilità dell’abbondanza di microrganismi, collettivamente indicati come microbiota, che si associano in simbiosi con i tessuti barriera del corpo esposti all’ambiente esterno – in particolare l’epidermide e la mucosa interna del tratto gastrointestinale, nonché i polmoni, il seno e il sistema genito-urinario.
Vi è un crescente riconoscimento del fatto che gli ecosistemi creati da batteri e funghi residenti – i microbiomi – hanno profondi effetti sulla salute e sulle malattie (87), una realizzazione guidata dalla capacità di selezionare le popolazioni di specie microbiche utilizzando tecnologie di sequenziamento e bioinformatica di prossima generazione. Per quanto riguarda il cancro, stanno diventando sempre più convincenti le prove che la variabilità polimorfica nei microbiomi tra gli individui di una popolazione può avere effetti profondi sui fenotipi del cancro (88, 89).
Studi di associazione su manipolazioni umane e sperimentali in modelli murini di cancro rivelano alcuni microrganismi, principalmente ma non esclusivamente batteri, che possono avere effetti protettivi o dannosi sullo sviluppo del cancro, sulla progressione maligna e sulla risposta alla terapia. Ciò vale anche per la complessità globale e la composizione del microbioma tissutale nel suo insieme. Sebbene il microbioma intestinale sia stato il pioniere di questa nuova frontiera, diversi tessuti e organi hanno microbiomi associati che mostrano caratteristiche distintive legate alle dinamiche della popolazione e alla diversità delle specie e sottospecie microbiche.
Questo crescente apprezzamento dell’importanza dei microbiomi polimorficamente variabili nella salute e nella malattia solleva la domanda: il microbioma è un tratto abilitante distinto che ha un ampio impatto, sia positivo che negativo, sull’acquisizione di capacità distintive per il cancro? Considererò questa possibilità di seguito e illustrerò le prove di alcuni dei microbiomi tissutali più importanti implicati nei tratti del cancro (Fig. 4), a partire dal microbioma più importante e apparentemente di maggior impatto, quello del tratto intestinale.
Figura 4
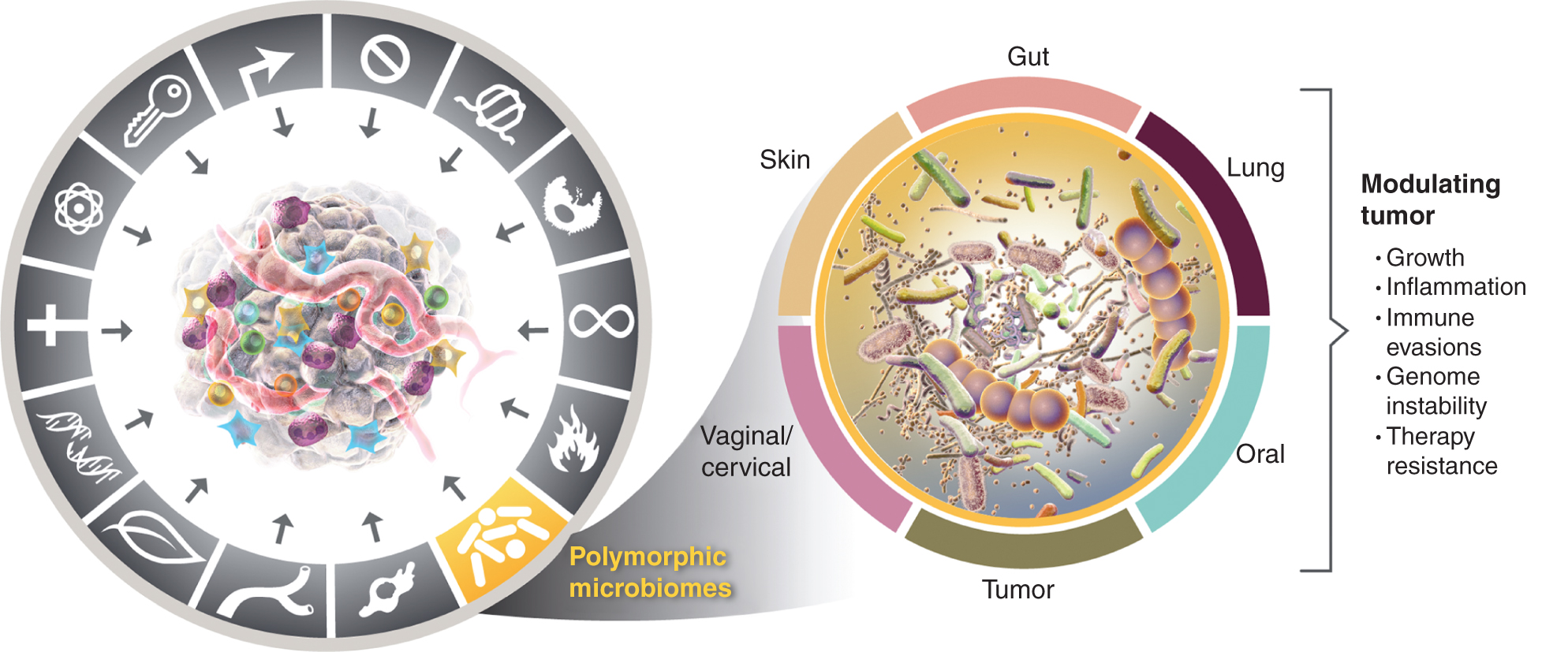
A sinistra, mentre le proprietà abilitanti dell’infiammazione che promuove il tumore e dell’instabilità genomica e della mutazione si sovrappongono, ci sono ragioni crescenti per concludere che i microbiomi polimorfici situati in un individuo rispetto a un altro nel colon, in altre membrane mucose e organi associati, o nei tumori stessi, possono influenzare molte delle abilità caratteristiche in vari modi – attraverso l’induzione o l’inibizione – e quindi possono essere una variabile strumentale e quasi indipendente nel puzzle di come il cancro si sviluppa, progredisce e risponde alla terapia. È vero, più microbiomi tissutali sono coinvolti nella modulazione dei fenotipi tumorali. Oltre al microbioma intestinale ampiamente studiato, altri microbiomi tissutali caratteristici e il microbioma tumorale sono coinvolti nella modulazione dell’acquisizione – sia positiva che negativa – delle capacità caratteristiche presentate in alcuni tipi di tumore. I marchi della grafica del cancro sono stati adottati da Hanahan e Weinberg (2).
Molteplici effetti modulatori del microbioma intestinale
È noto da tempo che il microbioma intestinale è fondamentale per la funzione dell’intestino crasso (colon) nel scomporre e importare i nutrienti nel corpo come parte dell’omeostasi metabolica, e che la disgregazione delle popolazioni microbiche – disbiosi – nel colon può causare uno spettro di malattie fisiologiche (87). Ciò include il sospetto che la predisposizione, lo sviluppo e la patogenesi del cancro del colon siano influenzati dal microbioma intestinale. Negli ultimi anni, convincenti studi funzionali utilizzando trapianti fecali da pazienti portatori di tumore del colon e da topi in topi riceventi predisposti allo sviluppo del cancro del colon hanno stabilito un principio: esistono microbiomi sia protettivi che promotori del cancro che coinvolgono specifiche specie batteriche che possono modulare l’insorgenza e la patogenesi dei tumori del colon (90).
I meccanismi attraverso i quali il microbiota conferisce questi ruoli modulatori sono ancora in fase di chiarimento, ma due effetti generali sono sempre più consolidati per i microbiomi che promuovono il tumore e, in alcuni casi, per specifiche specie batteriche che promuovono il tumore. Il primo effetto è la mutagenesi dell’epitelio del colon come risultato della produzione di tossine batteriche e altre molecole che danneggiano direttamente il DNA o interrompono i sistemi che mantengono l’integrità genomica o altrimenti stressano le cellule, influenzando indirettamente la fedeltà della replicazione e riparazione del DNA. Un tipico esempio è l'E. coli, che porta il locus PKS, che ha dimostrato di mutagenizzare il genoma umano ed è coinvolto nella trasmissione di mutazioni che abilitano il marchio (91).
Inoltre, è stato riportato che i batteri si legano alla superficie delle cellule epiteliali del colon e producono mimetici dei ligandi che stimolano la proliferazione epiteliale, contribuendo alla caratteristica capacità di segnalazione proliferativa delle cellule neoplastiche (88). Un altro meccanismo attraverso il quale tipi specifici di batteri promuovono lo sviluppo del tumore sono i batteri produttori di butirrato, la cui abbondanza è aumentata nei pazienti con cancro del colon-retto (92).
La produzione del metabolita butirrato ha effetti fisiologici complessi, inclusa l'induzione di cellule epiteliali e fibroblastiche senescenti. Un modello murino di carcinogenesi del colon colonizzato con batteri produttori di butirrato ha sviluppato più tumori rispetto ai topi privi di tali batteri; La connessione tra la senescenza indotta dal butirrato e l'aumento della tumorigenesi del colon è stata dimostrata attraverso l'uso di un farmaco senolitico che uccide le cellule senescenti, compromettendo la crescita del tumore (92).
Inoltre, il butirrato prodotto dai batteri ha effetti pleiotropici e paradossali sulle cellule differenziate rispetto alle cellule indifferenziate (staminali) nell'epitelio del colon in condizioni in cui la barriera intestinale è interrotta (disbiosi) e i batteri sono invasivi, influenzando, ad esempio, l'energia cellulare e il metabolismo, la modificazione degli istoni, la progressione del ciclo cellulare e l'infiammazione immunitaria innata (promotrice del tumore) che immunosopprime le risposte immunitarie adattative (93).
Infatti, un’ampia azione dei microbiomi polimorfici comporta la modulazione del sistema immunitario adattativo e innato attraverso diversi percorsi, inclusa la produzione di fattori “immunomodulatori” da parte di batteri che attivano sensori di danno sulle cellule immunitarie epiteliali o residenti, portando all’espressione di un repertorio diversificato di chemochine e citochine che possono modellare l’abbondanza e le proprietà delle cellule immunitarie che popolano l’epitelio del colon e lo stroma sottostante e i linfonodi drenanti.
Inoltre, alcuni batteri possono violare sia il biofilm protettivo che il muco che riveste l’epitelio del colon e distruggere le giunzioni strette cellula epiteliale che mantengono collettivamente l’integrità della barriera fisica che normalmente suddivide in compartimenti il microbioma intestinale. Dopo aver invaso lo stroma, i batteri possono innescare risposte immunitarie sia innate che adattative provocando la secrezione di un repertorio di citochine e chemochine. Una manifestazione potrebbe essere la creazione di microambienti immunitari che promuovono o antagonizzano il tumore, che di conseguenza proteggono o facilitano la tumorigenesi e la progressione maligna.
Di conseguenza, la modulazione dei parametri intrecciati di (i) l'induzione dell'infiammazione (innata) che promuove il tumore e (ii) la fuga dalla distruzione immunitaria (adattativa) da parte dei microbiomi caratteristici nei singoli pazienti può essere associata non solo alla prognosi ma anche alla risposta o alla resistenza alle immunoterapie con inibitori del checkpoint immunitario e ad altre modalità terapeutiche (Una manifestazione può essere la creazione di microambienti immunitari che promuovono o antagonizzano il tumore, che di conseguenza si verificano. Proteggere o facilitare lo sviluppo del tumore e progressione maligna.
Di conseguenza, la modulazione dei parametri intrecciati di (i) l'induzione dell'infiammazione (innata) che promuove il tumore e (ii) la fuga dalla distruzione immunitaria (adattativa) da parte dei microbiomi caratteristici nei singoli pazienti può essere associata non solo alla prognosi ma anche alla risposta o alla resistenza alle immunoterapie con inibitori del checkpoint immunitario e ad altre modalità terapeutiche (Una manifestazione può essere la creazione di microambienti immunitari che promuovono o antagonizzano il tumore, che di conseguenza si verificano proteggendo o facilitando lo sviluppo del tumore e progressione maligna).
Di conseguenza, la modulazione dei parametri intrecciati di (i) induzione dell'infiammazione (innata) che promuove il tumore e (ii) fuga dalla distruzione immunitaria (adattativa) da parte di microbiomi distintivi nei singoli pazienti può essere associata non solo alla prognosi ma anche alla risposta o alla resistenza alle immunoterapie con inibitori del checkpoint immunitario e altre modalità terapeutiche (89, 94-96). Una prova di concetto preliminare proviene da studi recenti che mostrano il ripristino dell’efficacia dell’immunoterapia dopo trapianti di microbiota fecale da pazienti che hanno risposto alla terapia in pazienti con melanoma che era progredito durante il precedente trattamento con blocco del checkpoint immunitario (97, 98).
I meccanismi molecolari attraverso i quali componenti distinti e variabili del microbioma intestinale modulano sistematicamente l’attività del sistema immunitario adattativo rimangono un mistero persistente, sia migliorando le risposte immunitarie antitumorali indotte dal blocco del checkpoint immunitario o, piuttosto, inducendo immunosoppressione sistemica o locale (intratumorale). Uno studio recente ha fatto luce: alcuni ceppi di Enterococcus (e altri batteri) esprimono una peptidoglicano idroliasi chiamata SagA, che rilascia mucopeptidi dalla parete batterica, che possono quindi circolare a livello sistemico e attivare il recettore del pattern NOD2, che a sua volta aumenta la risposta delle cellule T e l'efficacia dell'immunoterapia checkpoint (99).
Vengono identificate e valutate funzionalmente altre molecole immunoregolatrici prodotte da specifiche sottospecie batteriche, tra cui l'inosina prodotta dai batteri, un metabolita limitante la velocità dell'attività delle cellule T ( 100 ). Questi e altri esempi iniziano a delineare i meccanismi molecolari attraverso i quali i microbiomi polimorfici modulano indirettamente e sistematicamente l’immunobiologia tumorale, al di là delle risposte immunitarie che seguono le interazioni fisiche dirette dei batteri con il sistema immunitario (101, 102).
A parte i collegamenti causali con il cancro del colon e il melanoma, la capacità dimostrata del microbioma intestinale di suscitare l’espressione di chemochine e citochine immunomodulatorie che entrano nella circolazione sistemica è apparentemente in grado di influenzare la patogenesi del cancro e la risposta alle terapie in altri organi del corpo (94, 95).
Un esempio illuminante riguarda lo sviluppo di colangiocarcinomi nel fegato: la disbiosi intestinale consente l’ingresso e il trasporto di batteri e prodotti batterici attraverso la vena porta al fegato, dove il TLR4 espresso sugli epatociti viene attivato per indurre l’espressione della chemochina CXCL1, che recluta cellule mieloidi granulocitiche che esprimono CXCR2 (gMDSC) che servono a sopprimere le cellule killer naturali per eludere la distruzione immunitaria (103) e probabilmente trasmettere altre capacità distintive. (85). In quanto tale, il microbioma intestinale è chiaramente implicato come una caratteristica abilitante che può alternativamente facilitare o proteggere da più tumori.
Oltre l’intestino: implicazione di microbiomi distinti in altri tessuti barriera
Quasi tutti i tessuti e gli organi direttamente o indirettamente esposti all'ambiente esterno sono anche depositi di microrganismi commensali (104). A differenza dell’intestino, dove il ruolo simbiotico del microbioma nel metabolismo è ben riconosciuto, i ruoli normali e patogeni del microbiota residente in queste diverse sedi stanno ancora emergendo.
Esistono evidenti differenze organo/tessuto specifiche nella costituzione dei microbiomi associati nell'omeostasi, nell'invecchiamento e nel cancro, con specie e frequenze sovrapposte e distintive rispetto a quelle del colon ( 104 , 105 ). Inoltre, gli studi di associazione stanno fornendo prove sempre più evidenti che i microbiomi tissutali locali antagonizzanti/protettori del tumore rispetto a quelli che promuovono il tumore, simili al microbioma intestinale, possono modulare la suscettibilità e la patogenesi ai tumori umani che insorgono nei loro organi associati ( 106 – 109 ).
Influenza del microbiota intratumorale?
Infine, i patologi riconoscono da tempo che i batteri possono essere rilevati nei tumori solidi, un’osservazione che ora è stata confermata da sofisticate tecnologie di profilazione. Ad esempio, in uno studio su 1.526 tumori che comprendevano sette tipi di cancro umano (osso, cervello, mammella, polmone, melanoma, ovaio e pancreas), ciascun tipo era caratterizzato da un microbioma distintivo, situato in gran parte nelle cellule tumorali e nelle cellule immunitarie. All'interno di ciascun tipo di tumore, è stato dimostrato che le variazioni nel microbioma tumorale sono associate a caratteristiche clinico-patologiche (110).
Allo stesso modo, il microbiota è stato rilevato in modelli murini di cancro del polmone e del pancreas geneticamente modificati de novo, e la loro assenza nei topi privi di germi e/o la loro eliminazione con antibiotici può essere dimostrata in grado di compromettere la tumorigenesi, implicando funzionalmente il microbioma tumorale come precursore dell'infiammazione che promuove il tumore e della progressione maligna (111, 112).
Studi di associazione sull’adenocarcinoma duttale pancreatico umano e test funzionali tramite trapianto fecale in topi portatori di tumore hanno dimostrato che le variazioni nel microbioma tumorale – e nel microbioma intestinale associato – modulano i fenotipi del sistema immunitario e la sopravvivenza ( 113 ). Una sfida importante per il futuro sarà quella di estendere queste implicazioni ad altri tipi di tumore e di distinguere i contributi potenzialmente separabili della costituzione e della variazione del microbioma tumorale da quelli del microbioma intestinale (e del tessuto locale di origine), magari identificando specie microbiche specifiche che sono funzionalmente influenti in un sito o nell’altro.
Riepilogo
Le domande intriganti per il futuro includono se il microbiota che risiede in diversi tessuti o che popola neoplasie incipienti ha la capacità di contribuire o interrompere l’acquisizione di altre capacità distintive oltre l’immunomodulazione e la mutazione genomica, influenzando così lo sviluppo e la progressione del tumore. Esistono prove che alcune specie batteriche possono stimolare direttamente il segno distintivo della segnalazione proliferativa, ad esempio nell'epitelio del colon (88), e possono modulare la soppressione della crescita alterando l'attività soppressore del tumore in diversi compartimenti dell'intestino (114), mentre gli effetti diretti su altre capacità caratteristiche, come evitare la morte cellulare, innescare l'angiogenesi e stimolare l'invasione e le metastasi, rimangono poco chiari, così come la generalizzabilità di queste osservazioni a molteplici forme di cancro umano.
Indipendentemente da ciò, ci sono argomentazioni sempre più convincenti secondo cui la variazione polimorfica nei microbiomi dell’intestino e di altri organi rappresenta una caratteristica distintiva di attivazione per l’acquisizione di abilità distintive (Fig. 4), anche se si sovrappone e integra quelle dell’instabilità e della mutazione del genoma e dell’infiammazione che promuove il tumore.
Cellule senescenti
La senescenza cellulare è una forma tipicamente irreversibile di arresto proliferativo che probabilmente si è evoluta come meccanismo protettivo per mantenere l’omeostasi dei tessuti, apparentemente come un meccanismo complementare alla morte cellulare programmata che serve a inattivare e, a tempo debito, rimuovere le cellule malate, disfunzionali o altrimenti non necessarie. Oltre a interrompere il ciclo di divisione cellulare, il programma di senescenza produce cambiamenti nella morfologia e nel metabolismo cellulare e, più profondamente, l’attivazione di un fenotipo secretorio associato alla senescenza (SASP), che comporta il rilascio di una pletora di proteine bioattive, comprese le chemochine.
Citochine e proteasi, la cui identità dipende dal tipo di cellula e di tessuto da cui deriva una cellula senescente ( 115–117). La senescenza può essere indotta nelle cellule da una varietà di condizioni, inclusi stress microambientali come la carenza di nutrienti e danni al DNA, nonché danni agli organelli e alle infrastrutture cellulari e squilibri nelle reti di segnalazione cellulare (115, 117), tutti verificatisi nel contesto dell'aumento osservato della frequenza delle cellule senescenti in vari organi durante l'invecchiamento (118, 119).
La senescenza cellulare è stata a lungo considerata un meccanismo protettivo contro la neoplasia, causando la senescenza delle cellule tumorali (120). La maggior parte degli iniziatori del programma di senescenza sopra menzionati sono associati a tumori maligni, in particolare danni al DNA dovuti a un'iperproliferazione aberrante, la cosiddetta senescenza indotta da oncogeni a causa di segnalazione iperattivata e senescenza indotta dalla terapia a causa di danni cellulari e genomici causati da chemioterapia e radioterapia.
In effetti, ci sono esempi consolidati dei benefici protettivi della senescenza nel limitare la progressione maligna (118, 119). Al contrario, un numero crescente di prove dimostra esattamente il contrario: in determinati contesti, le cellule senescenti stimolano in modo differenziale lo sviluppo del tumore e la progressione maligna (119, 121).
In un caso di studio approfondito, le cellule senescenti nei topi anziani sono state ablate farmacologicamente, riducendo in modo specifico le cellule senescenti che esprimono caratteristicamente l'inibitore del ciclo cellulare p16 - INK4a: oltre a ritardare diversi sintomi legati all'età, ciò ha comportato una deplezione delle cellule senescenti nei topi anziani con una ridotta incidenza di tumorigenesi spontanea e morte associata al cancro (122).
Si ritiene che il meccanismo principale attraverso il quale le cellule senescenti promuovono i fenotipi tumorali sia il SASP, che ha dimostrato di essere in grado di mediare le molecole di segnalazione (e le proteasi che si attivano e/o disattivano) in modo paracrino per mediare le capacità tipiche. Pertanto, in vari sistemi sperimentali, è stato dimostrato che le cellule tumorali senescenti contribuiscono in vari modi alla segnalazione proliferativa, evitano l'apoptosi, inducono l'angiogenesi, stimolano l'invasione e le metastasi e sopprimono l'immunità tumorale (116, 118, 120, 121).
Ancora un altro aspetto degli effetti delle cellule tumorali senescenti sui fenotipi tumorali coinvolge stati cellulari senescenti transitori e reversibili, per cui le cellule tumorali senescenti possono sfuggire al loro stato non proliferativo di espressione di SASP e riprendere la proliferazione cellulare e la manifestazione delle capacità associate di cellule oncogene completamente vitali (44).
Tale senescenza transitoria è meglio documentata nei casi di resistenza alla terapia (44), che rappresenta una forma di quiescenza che elude il targeting terapeutico delle cellule tumorali proliferanti, ma può rivelarsi più ampiamente efficace in altri stadi di sviluppo del tumore, progressione maligna e metastasi.
Inoltre, le capacità di promozione dei segni distintivi delle cellule senescenti non si limitano alle cellule tumorali senescenti. È stato dimostrato che i fibroblasti associati al cancro (CAF) senescono nei tumori, dando origine a CAF senescenti che hanno dimostrato di promuovere il tumore conferendo capacità caratteristiche alle cellule tumorali nella TME (115, 116, 121).
Inoltre, i fibroblasti senescenti nei tessuti normali, formati in parte dall'invecchiamento naturale o dagli insulti ambientali, sono similmente coinvolti nel rimodellamento dei microambienti tissutali attraverso il loro SASP per fornire supporto paracrino per l'invasione locale (i cosiddetti "effetti di campo") e le metastasi a distanza (116) di neoplasie che si sviluppano nelle vicinanze.
Inoltre, è stato dimostrato che i fibroblasti senescenti nella pelle invecchiata reclutano, tramite il loro SASP, cellule immunitarie innate che sono sia immunosoppressori delle risposte immunitarie antitumorali adattive ancorate alle cellule T CD8 sia stimolano la crescita del tumore cutaneo (123), quest'ultimo effetto riflette forse i contributi paracrini di tali cellule immunitarie innate (cellule mieloidi, neutrofili e macrofagi) ad altre capacità caratteristiche.
Sebbene meno consolidato, sembra probabile che altre abbondanti cellule stromali che popolano specifici microambienti tumorali vadano incontro a senescenza, modulando così le caratteristiche del cancro e i risultanti fenotipi tumorali. Ad esempio, le cellule endoteliali tumorali senescenti indotte dalla terapia possono aumentare la proliferazione, l’invasione e le metastasi nei modelli di cancro al seno (124, 125).
Certamente, tali prove giustificano indagini in altri tipi di tumore per valutare la senescenza generale dei fibroblasti, delle cellule endoteliali e di altre cellule stromali come forza trainante nello sviluppo del tumore. Attualmente non sono chiari anche i meccanismi regolatori e i determinanti funzionali attraverso i quali un particolare tipo di cellula senescente in una particolare TME suscita un SASP che promuove il tumore rispetto a un SASP che lo antagonizza, che apparentemente può essere indotto alternativamente nello stesso tipo di cellula senescente, forse da diversi iniziatori quando immersi in caratteristici microambienti fisiologici e neoplastici.
Riepilogo
Il concetto che i tumori sono costituiti da cellule tumorali geneticamente trasformate che interagiscono e traggono beneficio dalle cellule accessorie (stromali) reclutate ed epigeneticamente/fenotipicamente corrotte è stato stabilito come cruciale per la patogenesi del cancro. Le considerazioni discusse sopra e descritte nelle revisioni e nei rapporti citati qui (e altrove) sostengono in modo convincente che le cellule senescenti (indipendentemente dall'origine cellulare) dovrebbero essere prese in considerazione per l'inclusione nell'elenco delle cellule funzionalmente significative nel microambiente tumorale (Fig. 5). Pertanto, le cellule senescenti dovrebbero essere prese in considerazione nella ricerca di una conoscenza approfondita dei meccanismi del cancro. Inoltre, il riconoscimento della loro importanza motiva l'obiettivo secondario di colpire terapeuticamente le cellule senescenti che promuovono il tumore di tutte le costituzioni, sia attraverso l'ablazione farmacologica o immunologica o riprogrammando il SASP in varianti antagonizzanti il tumore (115, 121, 126).
Figura 5

Sottotipi eterogenei di cellule tumorali e tipi e sottotipi di cellule stromali sono funzionalmente integrati nelle manifestazioni dei tumori come organi illegali. Prove crescenti suggeriscono che i derivati cellulari senescenti di molti di questi componenti cellulari della TME e dei loro SASP variabili sono coinvolti nella modulazione delle capacità distintive e dei conseguenti fenotipi tumorali. I marchi della grafica del cancro sono stati adottati da Hanahan e Weinberg (2).
Osservazioni conclusive
Mentre gli otto tratti distintivi del cancro e le loro due caratteristiche di supporto hanno dimostrato di avere un valore euristico duraturo nella concettualizzazione del cancro, le considerazioni presentate sopra suggeriscono che potrebbero esserci nuovi aspetti di una certa generalità e quindi importanti per una comprensione più completa delle complessità, dei meccanismi e delle manifestazioni della malattia. Applicando la metrica dell'indipendenza distinguibile, se non completa, dai 10 attributi fondamentali, è discutibile che questi quattro parametri - previa ulteriore convalida e generalizzazione oltre i casi di studio presentati - potrebbero essere integrati nei tratti distintivi dello schema del cancro (Fig. 6).
Pertanto, la plasticità cellulare potrebbe essere aggiunta all’elenco delle capacità principali. Sebbene l’ottavo nucleo e questa nuova abilità siano concettualmente distinguibili in base alla loro definizione di tratti distintivi, aspetti della loro regolamentazione sono almeno parzialmente collegati in alcuni e forse molti tumori. Ad esempio, molteplici segni distintivi sono modulati in modo coordinato da fattori oncogeni canonici in alcuni tipi di tumore, tra cui
- (I) KRAS ( https://cancer.sanger.ac.uk/cosmic/census-page/KRAS ),
- (II) MYC ( https://cancer.sanger.ac.uk/cosmic/census-page/MYC ),
- (III) NOTCH ( https://cancer.sanger.ac.uk/cosmic/census-page/NOTCH1 ; Ref. 127) und
- (IV) TP53 ( https://cancer.sanger.ac.uk/cosmic/census-page/TP53 )
Figura 6
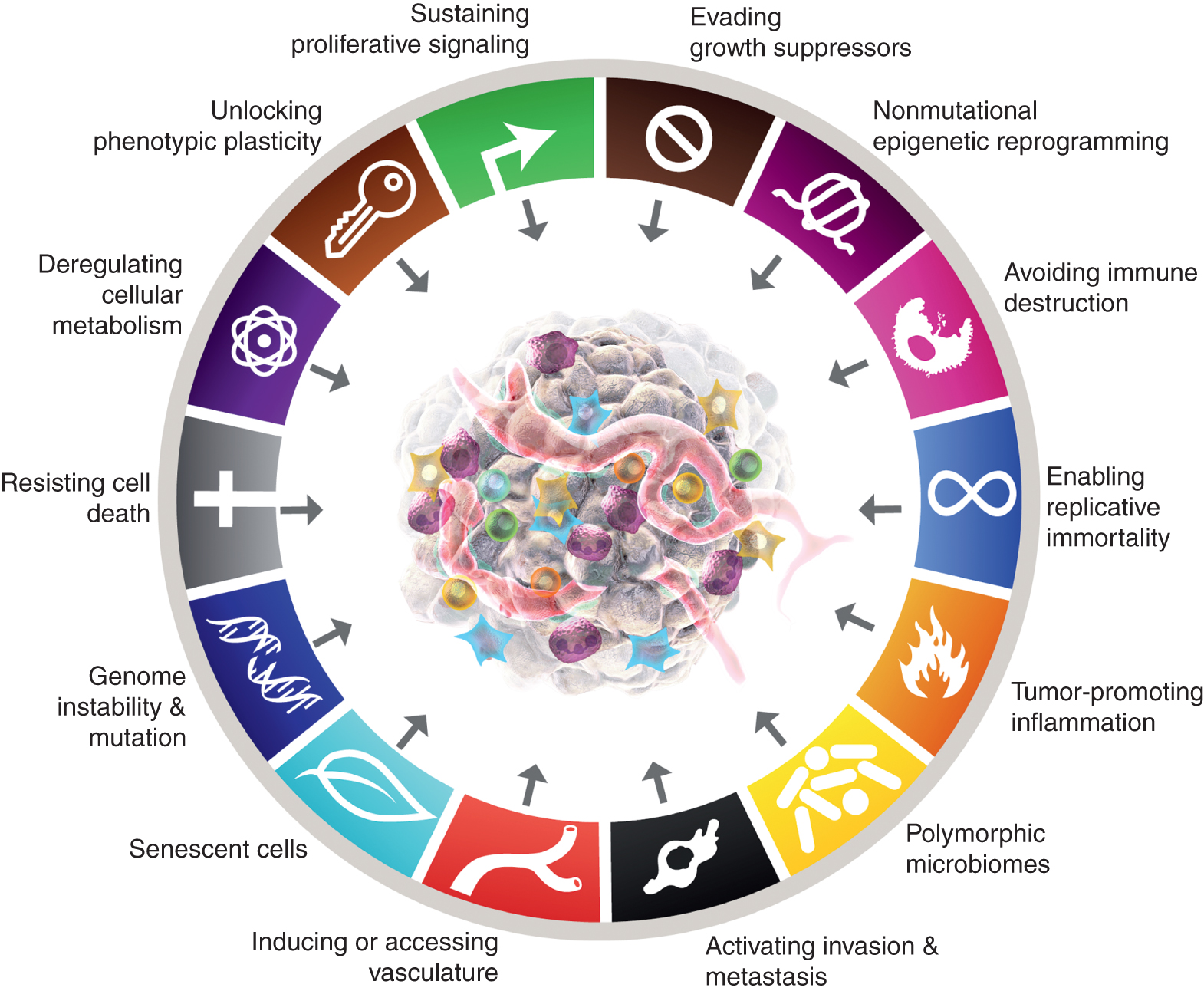
Vengono mostrate le nuove aggiunte canoniche e attese agli “Hallmarks of Cancer”. Questo articolo solleva la possibilità, con l’obiettivo di stimolare il dibattito, la discussione e l’elaborazione sperimentale, che alcuni o tutti i quattro nuovi parametri saranno riconosciuti come generici per molteplici forme di cancro umano e quindi adatti per l’integrazione nella concettualizzazione centrale delle caratteristiche del cancro. I marchi della grafica del cancro sono stati adottati da Hanahan e Weinberg (2).
Oltre ad aggiungere plasticità cellulare al roster, la riprogrammazione epigenetica non mutazionale e le variazioni polimorfiche possono essere integrate nei microbiomi di organi/tessuti come determinanti meccanicistici – proprietà abilitanti – attraverso i quali vengono acquisite capacità distintive, insieme all’infiammazione che promuove il tumore (a sua volta parzialmente interconnessa al microbioma), oltre le mutazioni e altre aberrazioni che manifestano i fattori oncogeni menzionati sopra.
Infine, cellule senescenti di diversa origine – comprese le cellule tumorali e varie cellule stromali – che contribuiscono funzionalmente allo sviluppo e alla progressione maligna del cancro, anche se in modi marcatamente diversi da quelli dei loro fratelli non senescenti, possono essere incluse come componenti generici della TME. In sintesi, si prevede che lo spiegamento di questi “palloncini sperimentali” preliminari stimolerà il dibattito, la discussione e ulteriori indagini sperimentali nella comunità di ricerca sul cancro sui parametri concettuali che definiscono la biologia, la genetica e la patogenesi del cancro.
Riferimenti
- Hanahan D , Weinberg RA . The hallmarks of cancer. Cell 2000;100:57–70.
- Hanahan D , Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Yuan S , Norgard RJ , Stanger BZ . Cellular plasticity in cancer. Cancer Discov 2019;9:837–51.
- Barker N , Ridgway RA , van Es JH , van de Wetering M , Begthel H , van den Born M et al . Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608–11.
- Perekatt AO , Shah PP , Cheung S , Jariwala N , Wu A , Gandhi V et al . SMAD4 suppresses WNT-driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res 2018;78:4878–90.
- Shih IM , Wang TL , Traverso G , Romans K , Hamilton SR , Ben-Sasson S et al . Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A 2001;98:2640–5.
- Ordóñez-Morán P , Dafflon C , Imajo M , Nishida E , Huelsken J . HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 2015;28:815–29.
- Tan SH , Barker N . Stemming colorectal cancer growth and metastasis: HOXA5 forces cancer stem cells to differentiate. Cancer Cell 2015;28:683–5.
- Köhler C , Nittner D , Rambow F , Radaelli E , Stanchi F , Vandamme N et al . Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 2017;21:679–93.
- Shah M , Bhoumik A , Goel V , Dewing A , Breitwieser W , Kluger H et al . A role for ATF2 in regulating MITF and melanoma development. PLoS Genet 2010;6:e1001258.
- Claps G , Cheli Y , Zhang T , Scortegagna M , Lau E , Kim H et al . A transcriptionally inactive ATF2 variant drives melanomagenesis. Cell Rep 2016;15:1884–92.
- Saghafinia S , Homicsko K , Di Domenico A , Wullschleger S , Perren A , Marinoni I et al . Cancer cells retrace a stepwise differentiation program during malignant progression. Cancer Discov 2021;11:2638–57.
- Yu X-X , Qiu W-L , Yang L , Zhang Y , He M-Y , Li L-C et al . Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J 2019;38:e100164.
- de Thé H . Differentiation therapy revisited. Nat Rev Cancer 2018;18:117–27.
- He LZ , Merghoub T , Pandolfi PP . In vivo analysis of the molecular pathogenesis of acute promyelocytic leukemia in the mouse and its therapeutic implications. Oncogene 1999;18:5278–92.
- Warrell RP , de Thé H , Wang ZY , Degos L . Acute promyelocytic leukemia. N Engl J Med 1993;329:177–89.
- Bots M , Verbrugge I , Martin BP , Salmon JM , Ghisi M , Baker A et al . Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014;123:1341–52.
- Ferrara FF , Fazi F , Bianchini A , Padula F , Gelmetti V , Minucci S et al . Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res 2001;61:2–7.
- Kaufman CK , Mosimann C , Fan ZP , Yang S , Thomas AJ , Ablain J et al . A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016;351:aad2197.
- Morris JP , Yashinskie JJ , Koche R , Chandwani R , Tian S , Chen C-C et al . α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019;573:595–9.
- Saha SK , Parachoniak CA , Ghanta KS , Fitamant J , Ross KN , Najem MS et al . Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110–4.
- Dang L , Su S-SM . Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem 2017;86:305–31.
- Waitkus MS , Diplas BH , Yan H . Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018;34:186–95.
- Phan TG , Croucher PI . The dormant cancer cell life cycle. Nat Rev Cancer 2020;20:398–411.
- Jiang M , Azevedo-Pouly AC , Deering TG , Hoang CQ , DiRenzo D , Hess DA et al . MIST1 and PTF1 collaborate in feed-forward regulatory loops that maintain the pancreatic acinar phenotype in adult mice. Mol Cell Biol 2016;36:2945–55.
- Krah NM , Narayanan SM , Yugawa DE , Straley JA , Wright CVE , MacDonald RJ et al . Prevention and reversion of pancreatic tumorigenesis through a differentiation-based mechanism. Dev Cell 2019;50:744–54.
- Krah NM , De La O J-P , Swift GH , Hoang CQ , Willet SG , Chen Pan F et al . The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015;4:e07125.
- Shi G , DiRenzo D , Qu C , Barney D , Miley D , Konieczny SF . Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 2013;32:1950–8.
- Kopp JL , von Figura G , Mayes E , Liu F-F , Dubois CL , Morris JP et al . Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50.
- Julian LM , McDonald AC , Stanford WL . Direct reprogramming with SOX factors: masters of cell fate. Curr Opin Genet Dev 2017;46:24–36.
- Grimm D , Bauer J , Wise P , Krüger M , Simonsen U , Wehland M et al . The role of SOX family members in solid tumours and metastasis. Semin Cancer Biol 2020;67:122–53.
- Mu P , Zhang Z , Benelli M , Karthaus WR , Hoover E , Chen C-C et al . SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84–8.
- Von Hoff DD , LoRusso PM , Rudin CM , Reddy JC , Yauch RL , Tibes R et al . Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72.
- Biehs B , Dijkgraaf GJP , Piskol R , Alicke B , Boumahdi S , Peale F et al . A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018;562:429–33.
- Boumahdi S , de Sauvage FJ . The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19:39–56.
- Groves SM , Ireland A , Liu Q , Simmons AJ , Lau K , Iams WT et al . Cancer Hallmarks Define a Continuum of Plastic Cell States between Small Cell Lung Cancer Archetypes [Internet]. Systems Biology; 2021 Jan. Available from: http://biorxiv.org/lookup/doi/10.1101/2021.01.22.427865.
- LaFave LM , Kartha VK , Ma S , Meli K , Del Priore I , Lareau C et al . Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 2020;38:212–28.
- Marjanovic ND , Hofree M , Chan JE , Canner D , Wu K , Trakala M et al . Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 2020;38:229–46.
- Drapkin BJ , Minna JD . Studying lineage plasticity one cell at a time. Cancer Cell 2020;38:150–2.
- Inoue Y , Nikolic A , Farnsworth D , Liu A , Ladanyi M , Somwar R et al . Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer [Internet]. Cancer Biology; 2020 Nov. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.11.12.368522.
- Dravis C , Chung C-Y , Lytle NK , Herrera-Valdez J , Luna G , Trejo CL et al . Epigenetic and transcriptomic profiling of mammary gland development and tumor models disclose regulators of cell state plasticity. Cancer Cell 2018;34:466–82.
- Malta TM , Sokolov A , Gentles AJ , Burzykowski T , Poisson L , Weinstein JN et al . Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–54.
- Miao Z-F , Lewis MA , Cho CJ , Adkins-Threats M , Park D , Brown JW et al . A dedicated evolutionarily conserved molecular network licenses differentiated cells to return to the cell cycle. Dev Cell 2020;55:178–94.
- De Blander H , Morel A-P , Senaratne AP , Ouzounova M , Puisieux A . Cellular plasticity: a route to senescence exit and tumorigenesis. Cancers 2021;13:4561.
- Merrell AJ , Stanger BZ . Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 2016;17:413–25.
- Baylin SB , Jones PA . Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 2016;8:a019505.
- Flavahan WA , Gaskell E , Bernstein BE . Epigenetic plasticity and the hallmarks of cancer. Science 2017;357:eaal2380.
- Jones PA , Issa J-PJ , Baylin S . Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630–41.
- Huang S . Tumor progression: Chance and necessity in Darwinian and Lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol 2012;110:69–86.
- Darwiche N . Epigenetic mechanisms and the hallmarks of cancer: an intimate affair. Am J Cancer Res 2020;10:1954–78.
- Feng Y , Liu X , Pauklin S . 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell 2021;12:440–54.
- Nam AS , Chaligne R , Landau DA . Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet 2021;22:3–18.
- Bitman-Lotan E , Orian A . Nuclear organization and regulation of the differentiated state. Cell Mol Life Sci CMLS 2021;78:3141–58.
- Goldberg AD , Allis CD , Bernstein E . Epigenetics: a landscape takes shape. Cell 2007;128:635–8.
- Zeng Y , Chen T . DNA methylation reprogramming during mammalian development. Genes 2019;10:257.
- Hegde AN , Smith SG . Recent developments in transcriptional and translational regulation underlying long-term synaptic plasticity and memory. Learn Mem 2019;26:307–17.
- Kim S , Kaang B-K . Epigenetic regulation and chromatin remodeling in learning and memory. Exp Mol Med 2017;49:e281.
- Thienpont B , Van Dyck L , Lambrechts D . Tumors smother their epigenome. Mol Cell Oncol 2016;3:e1240549.
- Gameiro PA , Struhl K . Nutrient deprivation elicits a transcriptional and translational inflammatory response coupled to decreased protein synthesis. Cell Rep 2018;24:1415–24.
- Lin GL , Monje M . Understanding the deadly silence of posterior fossa A ependymoma. Mol Cell 2020;78:999–1001.
- Michealraj KA , Kumar SA , Kim LJY , Cavalli FMG , Przelicki D , Wojcik JB et al . Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell 2020;181:1329–45.
- Bakir B , Chiarella AM , Pitarresi JR , Rustgi AK . EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol 2020;30:764–76.
- Gupta PB , Pastushenko I , Skibinski A , Blanpain C , Kuperwasser C . Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 2019;24:65–78.
- Lambert AW , Weinberg RA . Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer 2021;21:325–38.
- Lindner P , Paul S , Eckstein M , Hampel C , Muenzner JK , Erlenbach-Wuensch K et al . EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis 2020;11:147.
- Javaid S , Zhang J , Anderssen E , Black JC , Wittner BS , Tajima K et al . Dynamic chromatin modification sustains epithelial-mesenchymal transition following inducible expression of Snail-1. Cell Rep 2013;5:1679–89.
- Serrano-Gomez SJ , Maziveyi M , Alahari SK . Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 2016;15:18.
- Skrypek N , Goossens S , De Smedt E , Vandamme N , Berx G . Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet TIG 2017;33:943–59.
- Li L , Hanahan D . Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 2013;153:86–100.
- Li L , Zeng Q , Bhutkar A , Galván JA , Karamitopoulou E , Noordermeer D et al . GKAP acts as a genetic modulator of NMDAR signaling to govern invasive tumor growth. Cancer Cell 2018;33:736–51.
- Mohammadi H , Sahai E . Mechanisms and impact of altered tumour mechanics. Nat Cell Biol 2018;20:766–74.
- Odenthal J , Takes R , Friedl P . Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis 2016;37:1117–28.
- Torres CM , Biran A , Burney MJ , Patel H , Henser-Brownhill T , Cohen A-HS et al . The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 2016;353:aaf1644.
- Puram SV , Tirosh I , Parikh AS , Patel AP , Yizhak K , Gillespie S et al . Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171:1611–24.
- Kinker GS , Greenwald AC , Tal R , Orlova Z , Cuoco MS , McFarland JM et al . Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 2020;52:1208–18.
- Murtha M , Esteller M . Extraordinary cancer epigenomics: thinking outside the classical coding and promoter box. Trends Cancer 2016;2:572–84.
- Nebbioso A , Tambaro FP , Dell’Aversana C , Altucci L . Cancer epigenetics: moving forward. PLoS Genet 2018;14:e1007362.
- Tavernari D , Battistello E , Dheilly E , Petruzzella AS , Mina M , Sordet-Dessimoz J et al . Non-genetic evolution drives lung adenocarcinoma spatial heterogeneity and progression. Cancer Discov 2021;11:1490–507.
- Heyn H , Vidal E , Ferreira HJ , Vizoso M , Sayols S , Gomez A et al . Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 2016;17:11.
- Saghafinia S , Mina M , Riggi N , Hanahan D , Ciriello G . Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 2018;25:1066–80.
- Audia JE , Campbell RM . Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8:a019521.
- Corces MR , Granja JM , Shams S , Louie BH , Seoane JA , Zhou W et al . The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898.
- Esteve-Puig R , Bueno-Costa A , Esteller M . Writers, readers and erasers of RNA modifications in cancer. Cancer Lett 2020;474:127–37.
- Janin M , Coll-SanMartin L , Esteller M . Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer 2020;19:70.
- Hanahan D , Coussens LM . Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–22.
- Lu Z , Zou J , Li S , Topper MJ , Tao Y , Zhang H et al . Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020;579:284–90.
- Thomas S , Izard J , Walsh E , Batich K , Chongsathidkiet P , Clarke G et al . The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res 2017;77:1783–812.
- Dzutsev A , Badger JH , Perez-Chanona E , Roy S , Salcedo R , Smith CK et al . Microbes and cancer. Annu Rev Immunol 2017;35:199–228.
- Helmink BA , Khan MAW , Hermann A , Gopalakrishnan V , Wargo JA . The microbiome, cancer, and cancer therapy. Nat Med 2019;25:377–88.
- Sears CL , Garrett WS . Microbes, microbiota, and colon cancer. Cell Host Microbe 2014;15:317–28.
- Pleguezuelos-Manzano C , Puschhof J , Rosendahl Huber A , van Hoeck A , Wood HM , Nomburg J et al . Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020;580:269–73.
- Okumura S , Konishi Y , Narukawa M , Sugiura Y , Yoshimoto S , Arai Y et al . Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat Commun 2021;12:5674.
- Salvi PS , Cowles RA . Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in homeostasis and disease. Cells 2021;10:1775.
- Fessler J , Matson V , Gajewski TF . Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 2019;7:108.
- Gopalakrishnan V , Helmink BA , Spencer CN , Reuben A , Wargo JA . The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 2018;33:570–80.
- Zitvogel L , Ma Y , Raoult D , Kroemer G , Gajewski TF . The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–70.
- Baruch EN , Youngster I , Ben-Betzalel G , Ortenberg R , Lahat A , Katz L et al . Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021;371:602–9.
- Davar D , Dzutsev AK , McCulloch JA , Rodrigues RR , Chauvin J-M , Morrison RM et al . Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021;371:595–602.
- Griffin ME , Espinosa J , Becker JL , Luo J-D , Carroll TS , Jha JK et al . Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 2021;373:1040–6.
- Mager LF , Burkhard R , Pett N , Cooke NCA , Brown K , Ramay H et al . Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369:1481–9.
- Ansaldo E , Belkaid Y . How microbiota improve immunotherapy. Science 2021;373:966–7.
- Ansaldo E , Farley TK , Belkaid Y . Control of immunity by the microbiota. Annu Rev Immunol 2021;39:449–79.
- Zhang Q , Ma C , Duan Y , Heinrich B , Rosato U , Diggs LP et al . Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov 2021;11:1248–67.
- Ding T , Schloss PD . Dynamics and associations of microbial community types across the human body. Nature 2014;509:357–60.
- Byrd AL , Liu M , Fujimura KE , Lyalina S , Nagarkar DR , Charbit B et al . Gut microbiome stability and dynamics in healthy donors and patients with non-gastrointestinal cancers. J Exp Med 2021;218:e20200606.
- Healy CM , Moran GP . The microbiome and oral cancer: more questions than answers. Oral Oncol 2019;89:30-3.
- Swaney MH , Kalan LR . Living in your skin: microbes, molecules and mechanisms. Infect Immun 2021;89:e00695–20.
- Willis JR , Gabaldón T . The human oral microbiome in health and disease: from sequences to ecosystems. Microorganisms 2020;8:308.
- Xu J , Peng J-J , Yang W , Fu K , Zhang Y . Vaginal microbiomes and ovarian cancer: a review. Am J Cancer Res 2020;10:743–56.
- Nejman D , Livyatan I , Fuks G , Gavert N , Zwang Y , Geller LT et al . The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80.
- Jin C , Lagoudas GK , Zhao C , Bullman S , Bhutkar A , Hu B et al . Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019;176:998–1013.
- Pushalkar S , Hundeyin M , Daley D , Zambirinis CP , Kurz E , Mishra A et al . The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 2018;8:403–16.
- McAllister F , Khan MAW , Helmink B , Wargo JA . The tumor microbiome in pancreatic cancer: bacteria and beyond. Cancer Cell 2019;36:577–9.
- Kadosh E , Snir-Alkalay I , Venkatachalam A , May S , Lasry A , Elyada E et al . The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020;586:133–8.
- Birch J , Gil J . Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34:1565–76.
- Faget DV , Ren Q , Stewart SA . Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 2019;19:439–53.
- Gorgoulis V , Adams PD , Alimonti A , Bennett DC , Bischof O , Bishop C et al . Cellular senescence: defining a path forward. Cell 2019;179:813–27.
- He S , Sharpless NE . Senescence in health and disease. Cell 2017;169:1000–11.
- Kowald A , Passos JF , Kirkwood TBL . On the evolution of cellular senescence. Aging Cell 2020;19:e13270.
- Lee S , Schmitt CA . The dynamic nature of senescence in cancer. Nat Cell Biol 2019;21:94–101.
- Wang B , Kohli J , Demaria M . Senescent cells in cancer therapy: friends or foes? Trends Cancer 2020;6:838–57.
- Baker DJ , Childs BG , Durik M , Wijers ME , Sieben CJ , Zhong J et al . Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–9.
- Ruhland MK , Loza AJ , Capietto A-H , Luo X , Knolhoff BL , Flanagan KC et al . Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 2016;7:11762.
- Hwang HJ , Lee Y-R , Kang D , Lee HC , Seo HR , Ryu J-K et al . Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett 2020;490:100–10.
- Wang D , Xiao F , Feng Z , Li M , Kong L , Huang L et al . Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res 2020;22:103.
- Amor C , Feucht J , Leibold J , Ho Y-J , Zhu C , Alonso-Curbelo D et al . Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020;583:127–32.
- Aster JC , Pear WS , Blacklow SC . The varied roles of notch in cancer. Annu Rev Pathol 2017;12:245–75.
- Kuczynski EA , Vermeulen PB , Pezzella F , Kerbel RS , Reynolds AR . Vessel co-option in cancer. Nat Rev Clin Oncol 2019;16:469–93.
Google ScholarCrossref