 Suche
Suche
 Mein Konto
Mein Konto
Kreftens kjennetegn: Nye dimensjoner

Forord The Hallmarks of Cancer Conceptualization er et heuristisk verktøy for å destillere den enorme kompleksiteten til kreftfenotyper og genotyper til et foreløpig sett med underliggende prinsipper. Etter hvert som kunnskapen om kreftmekanismer har utviklet seg, har andre fasetter av sykdommen dukket opp som potensielle forbedringer. Dette øker utsiktene til at fenotypisk plastisitet og uordnet differensiering er en distinkt karakteristisk evne, og at ikke-mutasjons epigenetisk omprogrammering og polymorfe mikrobiomer begge representerer karakteristiske muliggjørende egenskaper som letter tilegnelsen av karakteristiske evner. Videre kan senescentceller av ulik opprinnelse legges til listen over funksjonelt viktige celletyper i tumormikromiljøet. Det betyr at kreft er skummelt i...

Kreftens kjennetegn: Nye dimensjoner
Forord
The Hallmarks of Cancer Conceptualization er et heuristisk verktøy for å destillere den enorme kompleksiteten til kreftfenotyper og genotyper til et foreløpig sett med underliggende prinsipper. Etter hvert som kunnskapen om kreftmekanismer har utviklet seg, har andre fasetter av sykdommen dukket opp som potensielle forbedringer. Dette øker utsiktene til at fenotypisk plastisitet og uordnet differensiering er en distinkt karakteristisk evne, og at ikke-mutasjons epigenetisk omprogrammering og polymorfe mikrobiomer begge representerer karakteristiske muliggjørende egenskaper som letter tilegnelsen av karakteristiske evner. Videre kan senescentceller av ulik opprinnelse legges til listen over funksjonelt viktige celletyper i tumormikromiljøet.
Betydning
Kreft er skremmende i bredden og omfanget av dets mangfold, som inkluderer genetikk, celle- og vevsbiologi, patologi og respons på terapi. Stadig kraftigere eksperimentelle og beregningsmessige verktøy og teknologier gir et skred av "store data" om de utallige sykdomsmanifestasjonene som kreft omfatter. Det integrerende konseptet som er nedfelt i kjennetegnene til kreft bidrar til å destillere denne kompleksiteten til en stadig mer logisk vitenskap, og de foreløpige nye dimensjonene som presenteres i dette perspektivet kan gi verdi til denne bestrebelsen for å bedre forstå mekanismene for karsinogenese og ondartet progresjon og anvende denne kunnskapen til kreftmedisin.
introduksjon
Kreftens kjennetegn er blitt foreslått som et sett med funksjonelle evner som menneskelige celler får når de beveger seg fra normalitet til neoplastiske veksttilstander, mer spesifikt evner som er kritiske for deres evne til å danne ondartede svulster. I disse artiklene ( 1, 2 ), Bob Weinberg og jeg listet opp det vi så for oss som fellestrekk som forener alle typer kreftceller på nivå med cellulær fenotype. Hensikten var å gi et konseptuelt rammeverk som ville tillate de komplekse fenotypene til forskjellige humane tumortyper og varianter å bli rasjonalisert i forhold til et felles sett av underliggende cellulære parametere. I utgangspunktet så vi for oss en komplementær inkludering av seks forskjellige merkefunksjoner og utvidet senere dette antallet til åtte.
Denne formuleringen ble påvirket av erkjennelsen av at kreft hos mennesker utvikler seg som produkter av flertrinnsprosesser og at tilegnelsen av disse funksjonelle egenskapene på en eller annen måte kan tilskrives de distinkte trinnene i tumorpatogenese. Mangfoldet av ondartet patogenese, som omfatter flere tumortyper og en økende mengde undertyper, involverer ulike aberrasjoner (og dermed ervervede evner og egenskaper) som er et resultat av vevsspesifikke barrierer som nødvendigvis omgås under visse tumorgenese-veier. Selv om vi erkjenner at slike spesialiserte mekanismer kan være nyttige, har vi begrenset betegnelsen av kjennetegnene til parametere som har en bred innvirkning på tvers av spekteret av kreft hos mennesker.
De åtte kjennetegnene inkluderer for tiden (fig.1, venstre) de ervervede evnene til å opprettholde proliferativ signalering, unngå vekstdempere, motstå celledød, muliggjøre replikativ udødelighet, indusere/tilgang kar, aktivere invasjon og metastaser, omprogrammere cellulær metabolisme og unngå ødeleggelse av immunsystemet. I den siste utviklingen av dette konseptet (2), ble deregulering av cellulær metabolisme og unngåelse av ødeleggelse av immunsystemet avgrenset som "fremvoksende kjennetegn", men nå, elleve år senere, er det tydelig at de, i likhet med de opprinnelige seks, kan betraktes som kjernekjennetegn ved kreft og er inkludert som sådan i den nåværende fortellingen (fig. 1, venstre).
Figur 1
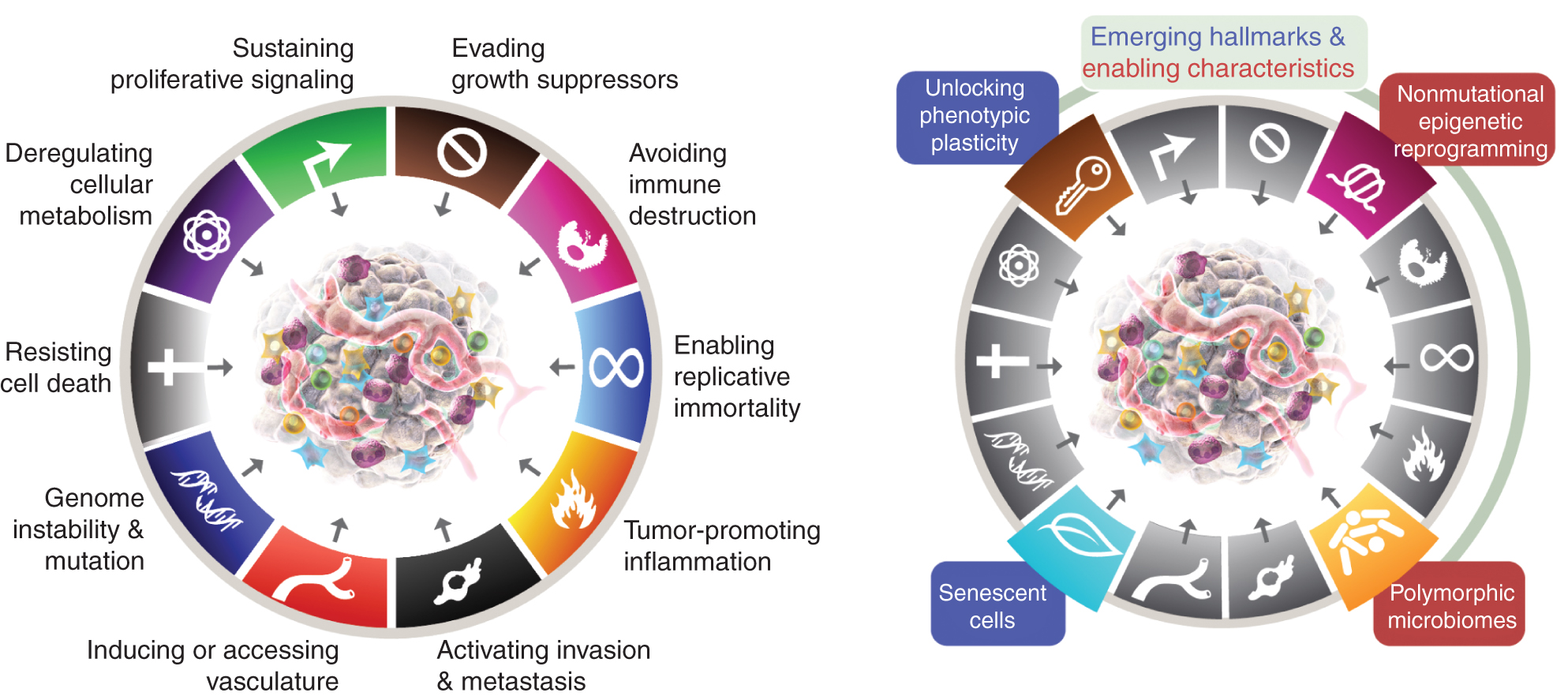
Kreftens kjennetegn omfatter for tiden åtte karakteristiske evner og to støttende egenskaper. I tillegg til de seks ervervede egenskapene - kjennetegn på kreft - som ble foreslått i 2000 (1), har de to foreløpige "emergent kjennetegnene" introdusert i 2011 (2) - cellulær energi (nå oftere referert til som "omprogrammering av cellulær metabolisme") og "unngå immundestruksjon" - blitt ansett som en del av kjernen som er tilstrekkelig validert.
Gitt den økende erkjennelsen av at svulster kan vaskulariseres adekvat, enten ved å slå på angiogenese eller ved å velge normal vevsvaskulatur (128), er dette kjennetegnet også bredere definert som evnen til å indusere eller på annen måte få tilgang til vaskulatur som støtter tumorvekst primært gjennom invasjon og metastasering.
2011-oppfølgeren inkluderte også "svulstfremmende betennelse" som en andre muliggjørende egenskap, som utfyller den overordnede "genomets ustabilitet og mutasjon", som sammen var fundamentalt involvert i å aktivere de åtte signatur-(funksjonelle) egenskapene som kreves for tumorvekst og -progresjon. Riktignok inkluderer denne anmeldelsen ytterligere foreslåtte nye kjennetegn og muliggjørende funksjoner, inkludert "å låse opp fenotypisk plastisitet", "ikke-mutasjons epigenetisk omprogrammering", "polymorfe mikrobiomer" og "aldrende celler." Varemerkene til kreftgrafikken ble adoptert fra Hanahan og Weinberg (2).
Som vi bemerket på den tiden, kan ikke disse særegne trekkene alene adressere kompleksiteten til kreftpatogenesen, dvs. de presise molekylære og cellulære mekanismene som gjør at de utviklende preneoplastiske cellene kan utvikle og tilegne seg disse avvikende fenotypiske evnene i løpet av tumorgenese og ondartet progresjon.
Følgelig har vi lagt til et annet konsept til diskusjonen presentert som "muliggjørende funksjoner", konsekvenser av den avvikende tilstanden til neoplasma som gir midler som kreftceller og svulster kan tilegne seg disse funksjonelle egenskapene. Som sådan gjenspeiles de muliggjørende egenskapene i molekylære og cellulære mekanismer som kjennetegn oppnås gjennom, snarere enn i selve de åtte ferdighetene ovenfor. Disse to aktiveringsprosessene var genom-ustabilitet og tumorfremmende betennelse.
Vi anerkjente videre at tumormikromiljøet (TME), definert her som sammensatt av heterogene og interaktive populasjoner av kreftceller og kreftstamceller sammen med en rekke rekrutterte stromale celletyper - det transformerte parenkymet og assosierte stroma - nå er allment verdsatt for å spille en viktig rolle i tumorgenese og ondartet progresjon.
Gitt den pågående interessen for disse formuleringene og vår fortsatte intensjon om å oppmuntre til kontinuerlig diskusjon og foredling av Hallmarks-skjemaet, er det hensiktsmessig å vurdere et ofte stilt spørsmål: Er det flere funksjoner ved denne konseptuelle modellen som kan inkorporeres, tatt i betraktning behovet for å sikre dette? at de er bredt anvendelige på tvers av spekteret av kreft hos mennesker? Følgelig presenterer jeg flere potensielle nye kjennetegn og muliggjørende funksjoner som etter hvert kan integreres som kjernekomponenter i kjennetegnene ved kreftkonseptualisering.
Disse parametrene er "å låse opp fenotypisk plastisitet", "ikke-mutasjons epigenetisk omprogrammering", "polymorfe mikrobiomer" og "senescent celler" (fig. 1, høyre). Det er viktig at eksemplene som presenteres til støtte for disse tesene er illustrerende, men på ingen måte omfattende, ettersom det er en voksende og stadig mer overbevisende mengde publiserte bevis som støtter hver vignett.
Tapping fenotypisk plastisitet
Under organogenese er utvikling, bestemmelse og organisering av celler i vev for å utføre homeostatiske funksjoner ledsaget av terminal differensiering, med stamceller som slutter å vokse, noen ganger irreversibelt, ettersom disse prosessene kulminerer. Som sådan er sluttresultatet av cellulær differensiering i de fleste tilfeller antiproliferativt, og danner en klar barriere for fortsatt spredning som er nødvendig for neoplasi.
Det er økende bevis på at det å låse opp den normalt begrensede kapasiteten for fenotypisk plastisitet til å omgå eller unnslippe tilstanden til terminal differensiering er en kritisk komponent i kreftpatogenesen ( 3 ). Denne plastisiteten kan virke i flere manifestasjoner (fig. 2). Således kan begynnende kreftceller som stammer fra en normal celle som har utviklet seg langs en bane som nærmer seg eller antar en fullstendig differensiert tilstand, reversere kurs ved å dedifferensiere tilbake til progenitorlignende celletilstander.
Omvendt kan neoplastiske celler som oppstår fra en progenitorcelle som er bestemt til å følge en vei som fører til terminal differensiering, kortslutte prosessen og opprettholde de ekspanderende kreftcellene i en delvis differensiert, progenitorlignende tilstand. Alternativt kan transdifferensiering forekomme der celler som opprinnelig var forpliktet til én differensieringsvei bytter til et helt annet utviklingsprogram og derved får vevsspesifikke egenskaper som ikke var forhåndsbestemt av deres normale opprinnelsesceller.
Følgende eksempler støtter argumentet om at forskjellige former for cellulær plastisitet avslører fenotypisk plastisitet. Til venstre er fenotypisk plastisitet uten tvil en ervervet karakteristisk evne som muliggjør ulike forstyrrelser av celledifferensiering, inkludert (i) dedifferensiering fra modne til stamceller, (ii) stoppet (terminal) differensiering fra stamceller og (iii) transdifferensiering til andre cellelinjer. Tre fremtredende moduser for svekket differensiering som er integrert i kreftpatogenesen er vist til høyre.
Ved å differensielt pervertere den normale differensieringen av stamceller til modne celler i utviklingslinjer, forenkles tumorgenese og ondartet progresjon som oppstår fra celler av opprinnelse i slike veier. Varemerkene til kreftgrafikken ble adoptert fra Hanahan og Weinberg (2).
Figur 2

Dedifferensiering
Kolonkarsinogenese er et eksempel på svekket differensiering, da det er et teleologisk behov for begynnende kreftceller for å unnslippe transportbåndet for terminal differensiering og eksfoliering, som i prinsippet kan skje gjennom dedifferensiering av tykktarmsepitelceller som ennå ikke har terminalt differensiert eller gjennom stoppet differensiering av disse celleforstammerne som gir forskjellig celle- og stamceller. celler. Både differensierte celler og stamceller har vært implisert som opprinnelsesceller for tykktarmskreft ( 4 – 6 ).
To utviklingsmessige transkripsjonsfaktorer (TF), homeobox-proteinet HOXA5 og SMAD4, sistnevnte involvert i BMP-signalering, er sterkt uttrykt i differensierende tykktarmsepitelceller og går vanligvis tapt i avanserte tykktarmskarsinomer, som karakteristisk uttrykker markører for stam- og stamceller. Funksjonelle forstyrrelser i musemodeller har vist at tvungen ekspresjon av HOXA5 i tykktarmskreftceller gjenoppretter differensieringsmarkører, undertrykker stamcellefenotyper og svekker invasjon og metastase, noe som gir en begrunnelse for dens karakteristiske nedregulering ( 7 , 8 ).
I kontrast fremtvinger SMAD4 både differensiering og undertrykkelse av spredning drevet av onkogen WNT-signalering, som avsløres av det konstruerte tapet av SMAD4-uttrykk, og gir en forklaring på tapet av uttrykk for å tillate dedifferensiering og deretter WNT-drevet hyperproliferasjon ( 5 ).
Spesielt er tapet av disse to "differensieringsundertrykkerne" med den resulterende dedifferensieringen assosiert med anskaffelse av andre kjennetegnsevner, så vel som andre kjennetegnsinduserende regulatorer, noe som kompliserer den strenge definisjonen av dette foreløpige kjennetegnet som separerbart og uavhengig.
En annen bevislinje gjelder det undertrykte uttrykket av MITF-mesterregulatoren for melanocyttdifferensiering, som ser ut til å være involvert i opprinnelsen til aggressive former for malignt melanom. Tap av denne utviklingsmessige TF er assosiert med reaktivering av neural crest progenitor-gener og nedregulering av gener som karakteriserer fullt differensierte melanocytter. Gjenopptredenen av nevrale kam-genene indikerer at disse cellene går tilbake til stamfadertilstanden hvorfra melanocytter oppstår utviklingsmessig.
Videre etablerte en linjesporingsstudie av BRAF-induserte melanomer modne pigmenterte melanocytter som opprinnelsesceller som gjennomgår dedifferensiering i løpet av tumorigenese ( 9 ). Spesielt induserer det mutante BRAF-onkogenet, funnet i mer enn halvparten av kutane melanomer, hyperproliferasjon, som går foran og kan derfor separeres mekanisk fra den påfølgende dedifferensieringen som oppstår fra nedregulering av MITF.
En annen studie impliserte funksjonelt oppreguleringen av utviklingsmessig TF ATF2, hvis karakteristiske uttrykk i mus og humane melanomer indirekte undertrykker MITF1, samtidig med ondartet progresjon av de følgelig dedifferensierte melanomcellene ( 10 ). Motsatt gir uttrykk i melanomer av mutante former av ATF2 som ikke kan undertrykke MITF godt differensierte melanomer ( 11 ).
Videre har en nylig studie ( 12 ) knyttet avstamningsdedifferensiering til ondartet progresjon av pankreasøycelle-neoplasmer til metastase-utsatte karsinomer; disse nevroendokrine cellene og avledede svulster oppstår fra en utviklingslinje som er forskjellig fra den som genererer det langt større antallet naboceller som danner den eksokrine og bukspyttkjertelen og de resulterende duktale adenokarsinomene.
Bemerkelsesverdig nok er flertrinnsdifferensieringsveien fra øy-stamceller til modne β-celler blitt grundig karakterisert ( 13 ). Sammenlignende transkriptomprofilering viser at adenomlignende øytumorer ligner mest på umodne, men differensierte insulinproduserende β-celler, mens de invasive karsinomene ligner mest på embryonale øycelleforløpere. Progresjon til dårlig differensierte karsinomer innebærer et innledende trinn med dedifferensiering, som i utgangspunktet ikke innebærer økt spredning eller redusert apoptose sammenlignet med godt differensierte adenomer, som begge har en tendens til å oppstå senere.
Dermed er det diskrete trinnet med dedifferensiering ikke drevet av observerbare endringer i de karakteristiske trekk ved vedvarende spredning og motstand mot apoptose. Snarere er oppreguleringen av et miRNA som tidligere var involvert i å spesifisere holmens stamfadertilstand en som nedreguleres under terminal differensiering av β-celler, 12).
Blokkert differensiering
Mens eksemplene ovenfor illustrerer hvordan undertrykkelse av differensieringsfaktoruttrykk kan lette tumorigenese ved å la bedre differensierte celler dedifferensiere til stamceller, kan i andre tilfeller ufullstendig differensierte stamceller lide av regulatoriske endringer som aktivt blokkerer deres videre progresjon til fullt differensierte, typisk ikke-proliferative tilstander.
Det har lenge vært dokumentert at akutt promyelocytisk leukemi (APL) er et resultat av en kromosomal translokasjon som smelter sammen PML-lokuset til genet som koder for retinsyre-α-kjernereseptoren (RARα). Myeloide stamceller som bærer slike translokasjoner er tilsynelatende ikke i stand til å fortsette sin vanlige terminale differensiering til granulocytter, noe som resulterer i celler fanget i et proliferativt, promyelocyttlignende stamfaderstadium ( 14 ).
Bevis på konseptet for denne ordningen kommer fra behandlingen av dyrkede APL-celler, musemodeller av sykdommen og berørte pasienter med retinsyre, liganden til RARα; Denne terapeutiske behandlingen får de neoplastiske APL-cellene til å differensiere til tilsynelatende modne, ikke-prolifererende granulocytter, og dermed kortslutte deres progressive proliferative ekspansjon (14–16).
En variant av dette temaet gjelder en annen form for akutt myeloid leukemi, denne som bærer t(8;21)-translokasjonen som produserer AML1-ETO-fusjonsproteinet. Dette proteinet alene kan transformere myeloide stamceller, i det minste delvis ved å blokkere deres differensiering. Terapeutisk intervensjon i musemodeller og pasienter med en farmakologisk hemmer av en kromatinmodifiserende histondeacetylase (HDAC) får myeloide leukemiceller til å gjenoppta differensiering til celler med en mer moden myeloid cellemorfologi. Følgende med denne reaksjonen er en reduksjon i proliferasjonskapasitet, og dermed svekke progresjonen av denne leukemien ( 17, 18 ).
Et tredje eksempel ved melanom involverer en utviklingsmessig TF, SOX10, som normalt nedreguleres under melanocyttdifferensiering. Gevinst og tap av funksjonsstudier i en sebrafiskmodell av BRAF-induserte melanomer har vist at unormalt opprettholdt ekspresjon av SOX10 blokkerer differensieringen av nevrale stamceller til melanocytter, og tillater dannelsen av BRAF-drevne melanomer ( 19 ).
Andre eksempler på differensieringsmodulatorer inkluderer metabolitten alfa-ketoglutarat (αKG), en nødvendig kofaktor for en rekke kromatinmodifiserende enzymer som har vist seg å være involvert i å stimulere visse differensierte celletilstander. Ved kreft i bukspyttkjertelen stimulerer tumorsuppressoren p53 produksjonen av αKG og opprettholdelsen av en mer differensiert celletilstand, mens et prototypisk tap av p53-funksjonen fører til reduksjon i αKG-nivåer og påfølgende dedifferensiering, som er assosiert med malign progresjon ( 20 ).
I en form for leverkreft resulterer ikke mutasjon av et isocitrat-dehydrogenase-gen (IDH1/2) i produksjon av differensieringsinduserende αKG, men snarere en beslektet "oncometabolite", D-2-hydroksigluterat (D2HG), som har vist seg å blokkere hepatocyttdifferensiering av leverprogenitorceller av forskjellig hepatocyttregulerende represjon av G-hepatitorceller gjennom D2-regulerende hepatitorceller. og ro, HNF4a.
D2HG-mediert undertrykkelse av HNF4a-funksjonen utløser en proliferativ utvidelse av hepatocytt-progenitorceller i leveren, som blir mottakelige for onkogen transformasjon ved påfølgende mutasjonsaktivering av KRAS-onkogenet, som driver malign progresjon til kolangiokarsinom i leveren ( 21 ). IDH1/2-mutanten og dens onkometabolitt D2HG fungerer også i en rekke myeloide og andre solide tumortyper, der D2HG hemmer αKG-avhengige dioksygenaser som kreves for histon- og DNA-metyleringshendelser som medierer endringer i kromatinstrukturen under utviklingsavstamningsdifferensiering, og dermed fryser utbruddet av kreftceller i a2 progeni2-tilstand (a2 progeni23).
Et ekstra, beslektet konsept er «bypassed differensiering», der delvis eller udifferensierte stamceller går ut av cellesyklusen og ligger i dvale i beskyttende nisjer, med potensial til å reinitiere proliferativ ekspansjon ( 24 ), men fortsatt med det selektive presset for å forstyrre deres programmerte differensiering på en eller annen måte.
Transdifferensiering
Konseptet med transdifferensiering har lenge vært anerkjent av patologer i form av vevsmetaplasi, der celler av en bestemt differensiert fenotype markant endrer sin morfologi for å bli klart gjenkjennelige som elementer i et annet vev, et fremtredende eksempel på dette er Barretts spiserør, hvor kronisk betennelse i den inflammatoriske epitheshagus-epituskvamus. transdifferensiering til et enkelt søyleepitel som er karakteristisk for tarmen, og letter dermed den påfølgende utviklingen av adenokarsinomer i stedet for plateepitelkarsinomene som forventes fra dette plateepitelet (3).
Nå avslører molekylære determinanter mekanismer for transdifferensiering i ulike kreftformer, både for tilfeller der grov vevsmetaplasi er åpenbar og andre der den er noe mer subtil, som følgende eksempler illustrerer.
Et informativt tilfelle for transdifferensiering som en diskret hendelse i tumorigenese gjelder pankreas duktalt adenokarsinom (PDAC), der en av de involverte cellene som kommer fra, pankreas acinarcellen, kan transdifferensiere til en duktal cellefenotype under initiering av neoplastisk utvikling. To TF-er – PTF1a og MIST1 – kontrollerer spesifikasjonen og vedlikeholdet av den differensierte acinære celletilstanden i bukspyttkjertelen via deres uttrykk i sammenheng med selvopprettholdende «feed-forward» regulatoriske looper ( 25 ).
Begge disse TF-ene nedreguleres ofte under neoplastisk utvikling og ondartet progresjon av PDAC fra mennesker og mus. Funksjonelle genetiske studier i mus og dyrkede humane PDAC-celler har vist at eksperimentelt tvungen ekspresjon av PTF1a svekker KRAS-indusert transdifferensiering og proliferasjon og kan også tvinge redifferensiering av allerede neoplastiske celler til en stillestående acinær celle-fenotype (26).
Motsatt utløser undertrykkelse av PTF1a-ekspresjon acinar-to-duct metaplasi, nemlig transdifferensiering, og sensibiliserer derved de kanallignende cellene for onkogen KRAS-transformasjon, og akselererer den påfølgende utviklingen av invasiv PDAC ( 27 ). På samme måte blokkerer tvungen ekspresjon av MIST1 i KRAS-uttrykkende bukspyttkjertel også transdifferensiering og svekker initieringen av bukspyttkjerteltumorigenese, som ellers forenkles av dannelsen av premaligne kanallignende (PanIN) lesjoner, mens genetisk sletting av MIST1 øker deres dannelse og initiering av 2KRAS-progresjon (initiering av neoplastisk 2KRAS).
Tap av enten PTF1- eller MIST1-ekspresjon under tumorigenese er assosiert med økt ekspresjon av en annen utviklingsregulerende TF, SOX9, som normalt er effektiv i spesifikasjon av ductal celle (27, 28). Tvunget oppregulering av SOX9, for derved å unngå behovet for nedregulering av PTF1a, og MIST1 har også vist seg å stimulere transdifferensieringen av acinarceller til en duktal cellefenotype som er følsom for KRAS-indusert neoplasi (29), noe som impliserer SOX9 som en nøkkelfunksjonell effektor i deres genese nedregulering av menneskelig PDAC.
Dermed kan tre TF-er som regulerer pankreatisk differensiering endres på forskjellige måter for å indusere en transdifferensiert tilstand som, i sammenheng med mutasjonsaktivering av KRAS, letter onkogen transformasjon og initiering av tumorigenese og ondartet progresjon.
Ytterligere medlemmer av SOX-familien av kromatinassosierte regulatoriske faktorer er på den ene siden i stor grad assosiert med både celleskjebnespesifikasjon og linjebytte i utvikling ( 30 ) og på den annen side med flere tumorassosierte fenotyper ( 31 ). Et annet fremtredende eksempel på SOX-mediert transdifferensiering involverer en mekanisme for terapeutisk resistens ved prostatakreft.
I dette tilfellet er tap av RB- og p53-tumorundertrykkere - hvis fravær er karakteristisk for nevroendokrine svulster - som svar på antiandrogenterapi er nødvendig, men ikke tilstrekkelig for den ofte observerte transformasjonen av godt differensierte prostatakreftceller til karsinomceller som har invadert differensieringslinjen med molekylære og histologiske trekk i celle- og nevroendoktrinene. androgen reseptor. I tillegg til tap av RB og p53, krever ervervet resistens mot antiandrogenterapi oppregulert ekspresjon av SOX2, et utviklingsregulerende gen, som har vist seg å bidra til å indusere transdifferensiering av de terapiresponsive adenokarsinomcellene til derivater som er i en nevroendokrin celletilstand som er motstandsdyktig mot terapi (32).
Et tredje eksempel viser også transdifferensiering som en strategi som brukes av karsinomceller for å unngå eliminering ved avstamningsspesifikk terapi, i dette tilfellet med basalcellekarsinomer (BCC) i huden behandlet med en farmakologisk hemmer av Hedgehog-Smoothened (HH/SMO) onkogen vei kjent for å drive den neoplastiske veksten av disse (33).
Legemiddelresistente kreftceller bytter til en utviklingsrelatert, men distinkt celletype via brede epigenetiske skift i spesifikke kromatindomener og endret tilgjengelighet for to superenhancers. Den nylig ervervede fenotypiske tilstanden til BCC-celler gjør at de kan opprettholde uttrykk for den onkogene WNT-signalveien, som igjen gir uavhengighet fra den medikament-undertrykte HH/SMO-signalveien (34).
Som forventet fra denne transdifferensieringen, skifter transkriptomet til kreftcellene fra en gensignatur som gjenspeiler den involverte opprinnelsescellen til BCC-er, nemlig hårsekkenes bule-stamceller, til en signatur som indikerer de basale stamcellene som befolker BCC-interfollikulær epidermis. Slik transdifferensiering for å muliggjøre medikamentresistens er i økende grad dokumentert ved ulike former for kreft ( 35 ).
Utviklingslinjeplastisitet ser også ut til å være utbredt i de viktigste undertypene av lungekarsinom, dvs. h. ved nevroendokrine karsinomer [småcellet lungekreft (SCLC)] og adenokarsinomer + plateepitelkarsinomer [kollektiv ikke-småcellet lungekreft (NSCLC)]. Enkeltcelle-RNA-sekvensering har avslørt bemerkelsesverdig dynamisk og heterogen konvertering mellom disse undertypene, samt markerte variasjoner deri, under stadiene av lungetumorigenese, påfølgende ondartet progresjon og respons på terapi (36 – 38).
Derfor, snarere enn den enkle konseptualiseringen av en ren klonal veksling fra en avstamning til en annen, maler disse studiene et mye mer komplekst bilde av dynamisk interkonverterende subpopulasjoner av kreftceller som viser trekk ved flere utviklingslinjer og differensieringsstadier, en nøktern innsikt i denne forbindelse for avstamningsbasert terapeutisk målretting av menneskelig lungekreft. Regulatoriske determinanter for denne dynamiske fenotypiske plastisiteten begynner å bli identifisert (37, 39, 40).
Sammendrag
De tre klassene av mekanismer beskrevet ovenfor fremhever selektive regulatorer av cellulær plastisitet som – i det minste delvis – kan skilles fra kjerne onkogene drivere og andre særegne evner. Utover disse eksemplene er det en betydelig mengde bevis som knytter mange former for kreft til nedsatt differensiering, som er ledsaget av anskaffelse av transkriptomsignaturer og andre fenotyper - for eksempel histologisk morfologi - som er assosiert med progenitor- eller stamcellestadier observert i det tilsvarende normale vevet. opprinnelse eller i andre mer fjernt beslektede celletyper og avstamninger ( 41 – 43 ).
Som sådan ser disse tre underklassene av fenotypisk plastisitet - dedifferensiering av modne celler tilbake til progenitortilstander, stoppet differensiering for å fryse utviklende celler i progenitor-/stamcelletilstander, og transdifferensiering til alternative cellelinjer - ut til å være effektive i flere krefttyper under primær tumorrespons, maligne progresjon og/eller terapi.
Det er imidlertid to konseptuelle hensyn. For det første er dedifferensiering og stoppet differensiering sannsynligvis sammenvevd, ettersom de ikke kan skilles fra hverandre i mange tumortyper der opprinnelsescellen - differensiert celle eller progenitor / stamcelle - enten er ukjent eller alternativt involvert. For det andre er ervervelsen eller vedlikeholdet av progenitorcellefenotyper og tapet av differensierte funksjoner, i de fleste tilfeller, en unøyaktig refleksjon av det normale utviklingsstadiet, fordypet i et miljø av andre karakteristiske endringer i kreftcellen som ikke er tilstede i naturlig utviklende celler.
Videre involverer en annen form for fenotypisk plastisitet cellulær senescens, diskutert mer generelt nedenfor, der kreftceller indusert til å gjennomgå tilsynelatende irreversibel aldring i stedet er i stand til å unnslippe og fortsette proliferativ ekspansjon (44). Til slutt, som med andre særegne evner, er ikke cellulær plastisitet en ny oppfinnelse eller aberrasjon av kreftceller, men snarere korrupsjon av latente, men aktiverbare evner som ulike normale celler bruker for å støtte homeostase, reparasjon og regenerering (45).
Samlet sett oppmuntrer disse illustrerende eksemplene til vurdering av at opplåsing av cellulær plastisitet for å muliggjøre ulike former for forstyrret differensiering representerer en distinkt særegen evne som skiller seg i regulering og cellulær fenotype fra de godt validerte kjernekjennetegnene til kreft (fig. 2).
Epigenetisk omprogrammering uten mutasjon
Den muliggjørende egenskapen til genom (DNA) ustabilitet og mutasjon er en grunnleggende komponent i kreftutvikling og patogenese. For tiden katalogiserer flere internasjonale konsortier mutasjoner gjennom genomet til humane kreftceller, i praktisk talt alle typer kreft hos mennesker, i ulike stadier av malign progresjon, inkludert metastatiske lesjoner, og under utviklingen av adaptiv terapiresistens. Et resultat er den nå utbredte erkjennelsen av at mutasjoner i gener som organiserer, modulerer og opprettholder kromatinarkitektur og dermed regulerer genuttrykk globalt, i økende grad blir oppdaget og funksjonelt knyttet til krefttrekk ( 46 – 48 ).
Videre er det argumenter for en annen tilsynelatende uavhengig form for genom-omprogrammering som involverer rent epigenetisk regulerte endringer i genuttrykk, en som kan kalles «ikke-mutasjons epigenetisk omprogrammering» (fig. 3). Faktisk ble tesen om mutasjonsløs kreftevolusjon og rent epigenetisk programmering av karakteristiske kreftfenotyper reist for nesten et tiår siden ( 49 ) og blir stadig mer diskutert ( 46, 50–52 ).
Figur 3

I likhet med det som skjer under embryogenese og vevsdifferensiering og homeostase, tyder akkumulerende bevis på at instrumentelle genregulerende kretsløp og nettverk i svulster kan kontrolleres av en mengde korrupte og co-opterte mekanismer som er uavhengige av genomets ustabilitet og genmutasjon. Varemerkene til kreftgrafikken ble adoptert fra Hanahan og Weinberg (2).
Selvfølgelig er begrepet ikke-mutasjons epigenetisk regulering av genuttrykk godt etablert som den sentrale mekanismen som medierer embryonal utvikling, differensiering og organogenese (53-55). Hos voksne involverer for eksempel langtidshukommelse endringer i gen- og histonmodifikasjon, i kromatinstruktur og i utløsning av genekspresjonsbrytere, som opprettholdes stabilt over tid av positive og negative tilbakemeldingssløyfer ( 56, 57 ). Økende bevis støtter ideen om at analoge epigenetiske endringer kan bidra til tilegnelse av karakteristiske evner under tumorutvikling og ondartet progresjon. For å støtte denne hypotesen er noen eksempler presentert nedenfor.
Mikromiljømekanismer for epigenetisk omprogrammering
Hvis ikke gjennom onkogene mutasjoner alene, hvordan omprogrammeres kreftcellegenomet? En voksende mengde bevis tyder på at de avvikende fysiske egenskapene til tumormikromiljøet kan forårsake brede endringer i epigenomet, hvorav endringer som er gunstige for fenotypisk seleksjon av egenskapsevner kan føre til klonal utvekst av kreftceller med forbedret egnethet for proliferativ ekspansjon.
Et vanlig trekk ved svulster (eller regioner i svulster) er hypoksi som følge av utilstrekkelig vaskularisering. Hypoksi reduserer for eksempel aktiviteten til TET-demetylaser, noe som fører til betydelige endringer i metylomet, spesielt hypermetylering ( 58 ). Utilstrekkelig vaskularisering vil sannsynligvis også begrense biotilgjengeligheten av kritiske blodbårne næringsstoffer, og næringsdeprivasjon har for eksempel vist seg å endre translasjonskontrollen og følgelig øke den ondartede fenotypen av brystkreftceller ( 59 ).
Et overbevisende eksempel på hypoksi-mediert epigenetisk regulering er en form for det alltid fatale pediatriske ependymomet. Som mange embryonale og pediatriske svulster, mangler denne formen tilbakevendende mutasjoner, spesielt mangel på drivermutasjoner i onkogener og tumorsuppressorer. Snarere har den unormale veksten av disse kreftcellene vist seg å være kontrollert av et hypoksi-indusert genreguleringsprogram ( 60, 61 ). Bemerkelsesverdig nok ligger den antatte opprinnelsescellen til denne kreften i et hypoksisk rom og sensibiliserer sannsynligvis cellene i den for å starte tumorgenese gjennom hittil ukjente kofaktorer.
Et annet overbevisende bevis for mikromiljø-mediert epigenetisk regulering gjelder den invasive vekstevnen til kreftceller. Et klassisk eksempel er reversibel induksjon av invasivitet av kreftceller i kantene av mange solide svulster, orkestrert av utviklingsreguleringsprogrammet kjent som epitel-til-mesenkymal overgang (EMT; ref. 62–64). Spesielt ble en masterregulator av EMT, ZEB1, nylig vist å indusere ekspresjonen av en histonmetyltransferase, SETD1B, som igjen opprettholder ZEB1-uttrykk i en positiv tilbakemeldingssløyfe som opprettholder den (invasive) regulatoriske tilstanden til EMT (65).
En tidligere studie dokumenterte tilsvarende at induksjon av EMT gjennom oppregulert ekspresjon av en beslektet TF, SNAIL1, forårsaket markante endringer i kromatinlandskapet som følge av induksjon av en rekke kromatinmodifikatorer hvis aktivitet viste seg å være nødvendig for opprettholdelse av den fenotypiske tilstanden ( 66 ). Videre kan en rekke tilstander og faktorer som oppleves av kreftceller i kantene av svulster, inkludert hypoksi og cytokiner utskilt av stromaceller, tilsynelatende indusere EMT og dermed invasivitet (67, 68).
Et slående eksempel på programmering av invasivitet av mikromiljøet, som angivelig ikke er relatert til EMT-programmet, involverer autokrin aktivering av en nevronal signalkrets som involverer utskilt glutamat og dets reseptor NMDAR (69, 70). Bemerkelsesverdig nok har den prototypiske stivheten til mange solide svulster, nedfelt i omfattende endringer i den ekstracellulære matrisen (ECM) som omslutter cellene i dem, dype implikasjoner for de invasive og andre fenotypiske egenskapene til kreftceller.
Sammenlignet med normal vevs-ECM som svulster oppstår fra, er tumor-ECM typisk preget av økt tverrbinding og tetthet, enzymatiske modifikasjoner og endret molekylær sammensetning som kollektivt orkestrerer, delvis gjennom integrinreseptorer for ECM-motiver, stivhetsindusert signalering og genuttrykksnettverk som induserer invasive 1 karakteristiske egenskaper (7).
I tillegg til slike reguleringsmekanismer som er utstyrt med det fysiske tumormikromiljøet, kan parakrin signalering, som omfatter løselige faktorer frigitt til det ekstracellulære miljøet av de ulike celletypene som befolker solide svulster, også bidra til induksjon av flere morfologisk distinkte invasive vekstprogrammer (72), hvorav bare ett - kalt "mesent" - kalt "mesent" ovenfor. reguleringsmekanisme for EMT.
Epigenetisk regulatorisk heterogenitet
En voksende kunnskapsbase øker forståelsen for viktigheten av intratumoral heterogenitet for å generere det fenotypiske mangfoldet der de mest egnede cellene for proliferativ ekspansjon og invasjon vokser ut av sine brødre og derfor velges for ondartet progresjon. En faset av denne fenotypiske heterogeniteten skyldes absolutt kronisk eller episodisk genomisk ustabilitet og resulterende genetisk heterogenitet i cellene som befolker en svulst.
Videre blir det stadig tydeligere at ikke-mutasjonsbasert epigenetisk heterogenitet kan eksistere. Et fremtredende eksempel er linker-histonet H1.0, som er dynamisk uttrykt og undertrykt i subpopulasjoner av kreftceller innenfor en rekke tumortyper, med påfølgende sekvestrering eller tilgjengelighet av megabasestore domener [73]. Spesielt ble populasjonen av kreftceller med undertrykt H1.0 funnet å vise stammelignende egenskaper, forbedret tumor-initierende evne og en assosiasjon med dårlig prognose hos pasienter.
Et annet eksempel på epigenetisk regulert plastisitet er beskrevet i humane orale plateepitelkarsinomer (SCC), hvor kreftceller ved de invasive marginene inntar en delvis EMT-tilstand (p-EMT) som mangler de nevnte mesenkymale TF-ene, men uttrykker andre EMT-definerende gener som ikke uttrykkes i den sentrale kjernen av svulstene (74).
p-EMT-cellene representerer åpenbart ikke klonal kompartmentalisering av mutasjonsforandrede celler: kulturer av primære tumoravledede kreftceller inneholder dynamiske blandinger av både p-EMT hi og p-EMT lo-celler, og når p-EMT hi/lo-celler ble FACS-renset og dyrket, ble begge gjenopprettet til blandede populasjoner på p-EMT i løpet av p-EMT-dager. Selv om parakrine signaler fra det tilstøtende stroma kan betraktes som deterministiske for p-EMT hi-tilstanden, argumenterer den stabile tilstedeværelsen og regenereringen av de to epigenetiske tilstandene i kultur for en kreftcelle-iboende mekanisme. Spesielt støttes denne konklusjonen av analysen av 198 cellelinjer som representerer 22 krefttyper, inkludert SCC, hvor 12 stabilt heterogene epigenetiske tilstander (inkludert p-EMT i SCC) ble påvist på forskjellige måter i cellelinjemodellene så vel som deres relaterte primærtumorer (75).
Igjen, de heterogene fenotypiske tilstandene kunne ikke knyttes til påvisbare genetiske forskjeller, og i flere tilfeller har FACS-sorterte celler i en bestemt tilstand vist seg å dynamisk re-ekvilibrere ved kultur, og rekapitulere en stabil likevekt mellom de heterogene tilstandene observert i de opprinnelige cellelinjene.
Videre belyser teknologier for genomomfattende profilering av ulike attributter – utover DNA-sekvensen og dens mutasjonsvariasjon – innflytelsesrike elementer i merknaden og organiseringen av kreftcellegenomet som korrelerer med pasientens prognose og i økende grad med karakteristiske evner (76 – 78). Epigenomisk heterogenitet blir avslørt av stadig kraftigere teknologier for profilering av genomomfattende DNA-metylering (79, 80), histonmodifikasjon (81), kromatintilgjengelighet (82) og post-transkripsjonell modifikasjon og translasjon av RNA (83, 84).
En utfordring med hensyn til postulatet som vurderes her, vil være å bestemme hvilke epigenomiske modifikasjoner i visse krefttyper (i) som har regulatorisk betydning og (ii) som er representative for ren ikke-mutasjonell omprogrammering, i motsetning til mutasjonsdrevet og dermed genomforklarlig ustabilitet.
Epigenetisk regulering av stromale celletyper som befolker tumormikromiljøet
Generelt antas det at hjelpecellene i tumormikromiljøet som funksjonelt bidrar til tilegnelsen av karakteristiske evner ikke lider av genetisk ustabilitet og mutasjonsomprogrammering for å forbedre deres tumorfremmende aktiviteter; snarere konkluderes det med at disse cellene – kreftassosierte fibroblaster, medfødte immunceller og endotelceller og pericytter i tumorvaskulaturen – blir epigenetisk omprogrammert ved rekruttering av løselige og fysiske faktorer som definerer det solide tumormikromiljøet ( 2 , 85 ).
Det forventes at de multi-omiske profileringsteknologiene som for tiden brukes på kreftceller i økende grad vil bli brukt til å studere tilleggscellene (stromale) i svulster for å belyse hvordan normale celler er skadet for funksjonelt å støtte tumorutvikling og progresjon. For eksempel antyder en fersk studie (86) at slik omprogrammering kan innebære modifikasjoner av epigenomet, i tillegg til induktiv utveksling av cytokiner, kjemokiner og vekstfaktorer som endrer intracellulære signalnettverk i alle disse celletypene:
Når musemodeller med lungemetastaser ble behandlet med en kombinasjon av en DNA-metyltransferasehemmer (5-azacytidin) og en histonmodifikasjonshemmer (en HDAC), ble de infiltrerende myeloidcellene funnet å ha gått over fra en umoden (svulstfremmende) stamfadertilstand til celler som ligner modne interstitial-lignende motparter (som) i ubehandlede svulster, var ikke i stand til å støtte de typiske egenskapene som kreves for effektiv metastatisk kolonisering ( 86). Det er tenkelig at multiomisk profilering og farmakologiske forstyrrelser vil tjene til å belyse den omprogrammerte epigenetiske tilstanden i slike myeloide celler, så vel som andre karakteristiske tilbehørscelletyper som befolker tumormikromiljøer.
Sammendrag
Til sammen støtter disse illustrerende øyeblikksbildene tesen om at epigenetisk omprogrammering uten mutasjon vil bli akseptert som en sann muliggjørende egenskap som tjener til å lette tilegnelsen av karakteristiske evner (fig. 3), forskjellig fra genomisk DNA-ustabilitet og mutasjon. Spesielt kan ikke-mutasjonell epigenetisk omprogrammering forventes å vise seg å være integrert for å muliggjøre den foreløpige nye særegne evnen til fenotypisk plastisitet diskutert ovenfor, spesielt som en drivkraft i den dynamiske transkriptomiske heterogeniteten som blir stadig mer godt dokumentert i ondartede kreftceller TME. Fremme av encellede multi-omiske profileringsteknologier vil kaste lys over de respektive bidragene og samspillet mellom mutasjonsdrevet og ikke-mutasjonsdrevet epigenetisk regulering i utviklingen av svulster under ondartet progresjon og metastase.
Polymorfe mikrobiomer
En vidtrekkende grense innen biomedisin utfolder seg ved å belyse mangfoldet og variasjonen til overfloden av mikroorganismer, samlet referert til som mikrobiota, som assosieres symbiotisk med kroppens barrierevev som er utsatt for det ytre miljøet - spesielt epidermis og indre slimhinner i mage-tarmsystemet, i tillegg til bryst og lunge.
Det er økende erkjennelse av at økosystemene skapt av fastboende bakterier og sopp – mikrobiomene – har dype effekter på helse og sykdom (87), en erkjennelse drevet av evnen til å screene populasjonene av mikrobielle arter ved hjelp av neste generasjons sekvenserings- og bioinformatikkteknologier. For kreft blir bevis stadig mer overbevisende for at polymorf variasjon i mikrobiomene mellom individer i en populasjon kan ha dype effekter på kreftfenotyper (88, 89).
Assosiasjonsstudier i menneskelige og eksperimentelle manipulasjoner i musemodeller av kreft avslører visse mikroorganismer, primært men ikke utelukkende bakterier, som kan ha enten beskyttende eller skadelige effekter på kreftutvikling, ondartet progresjon og respons på terapi. Dette gjelder også den globale kompleksiteten og sammensetningen av et vevsmikrobiom som helhet. Mens tarmmikrobiomet var pioneren for denne nye grensen, har flere vev og organer assosiert mikrobiomer som viser karakteristiske trekk relatert til populasjonsdynamikk og mangfoldet av mikrobielle arter og underarter.
Denne økende forståelsen av viktigheten av polymorfisk variable mikrobiomer i helse og sykdom reiser spørsmålet: Er mikrobiomet en distinkt muliggjørende egenskap som i stor grad påvirker, både positivt og negativt, tilegnelsen av særegne evner for kreft? Jeg vurderer denne muligheten nedenfor og illustrerer bevis for noen av de fremtredende vevsmikrobiomene som er involvert i krefttrekk (fig. 4), og starter med det mest fremtredende og tilsynelatende mest virkningsfulle mikrobiomet, det i tarmkanalen.
Figur 4
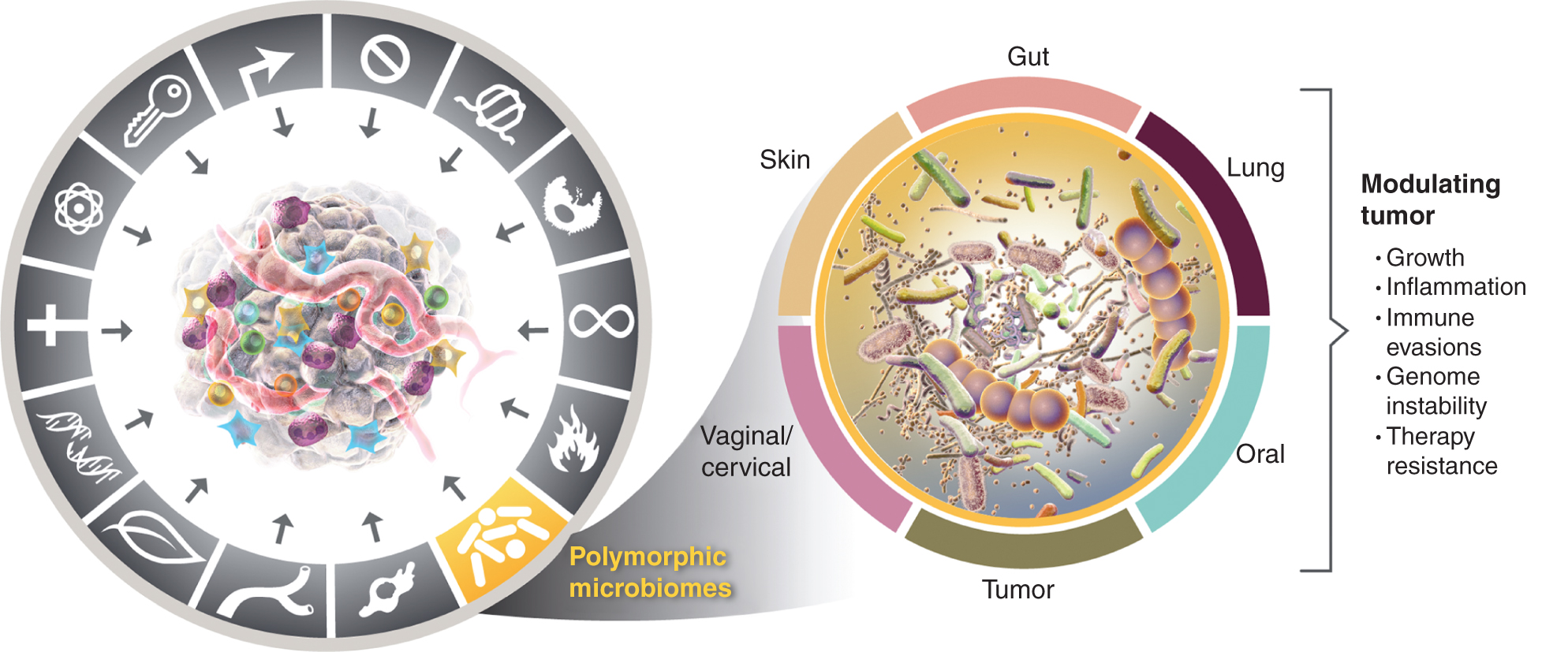
Til venstre, mens de muliggjørende egenskapene til svulstfremmende betennelse og genomisk ustabilitet og mutasjon overlapper hverandre, er det økende grunn til å konkludere med at polymorfe mikrobiomer lokalisert i ett individ sammenlignet med et annet i tykktarmen, i andre slimhinner og tilknyttede organer, eller i selve svulstene, kan påvirke mange av de karakteristiske evnene på en rekke måter - enten gjennom induksjon og inhibering eller kan derfor være en variabel inhibering eller inhibering. puslespill om hvordan kreft utvikler seg, utvikler seg og vokser reagerer på terapi. Riktignok er flere vevsmikrobiomer involvert i å modulere tumorfenotyper. I tillegg til det mye studerte tarmmikrobiomet, er andre karakteristiske vevsmikrobiomer så vel som tumormikrobiomet involvert i å modulere tilegnelsen - både positiv og negativ - av de karakteristiske evnene som presenteres i visse tumortyper. Varemerkene til kreftgrafikken ble adoptert fra Hanahan og Weinberg (2).
Flere modulerende effekter av tarmmikrobiomet
Det har lenge vært kjent at tarmmikrobiomet er grunnleggende for funksjonen til tykktarmen (tykktarmen) i å bryte ned og importere næringsstoffer til kroppen som en del av metabolsk homeostase, og at forstyrrelse av mikrobielle populasjoner – dysbiose – i tykktarmen kan forårsake et spekter av fysiologiske sykdommer (87). Dette inkluderer mistanke om at mottakelighet, utvikling og patogenesen av tykktarmskreft påvirkes av tarmmikrobiomet. De siste årene har overbevisende funksjonelle studier som bruker fekale transplantasjoner fra pasienter som bærer tykktarmssvulster og mus til mottakermus som er disponert for utvikling av tykktarmskreft etablert et prinsipp: det finnes både kreftbeskyttende og svulstfremmende mikrobiomer som involverer spesifikke bakteriearter som kan modulere forekomsten og patogenesen av tykktarmssvulster (90).
Mekanismene som mikrobiota gir disse modulerende rollene blir fortsatt belyst, men to generelle effekter er stadig mer etablert for tumorfremmende mikrobiomer og, i noen tilfeller, for spesifikke tumorfremmende bakteriearter. Den første effekten er mutagenese av tykktarmsepitelet som et resultat av produksjonen av bakterielle toksiner og andre molekyler som enten direkte skader DNA eller forstyrrer systemene som opprettholder genomisk integritet eller på annen måte stresser celler, og indirekte påvirker trofastheten til DNA-replikasjon og reparasjon. Et typisk eksempel er E. coli, som bærer PKS-lokuset, som har vist seg å mutagenisere det menneskelige genomet og er involvert i overføring av mutasjoner som muliggjør merket (91).
Videre er det rapportert at bakterier binder seg til overflaten av tykktarmsepitelceller og produserer ligandmimetikk som stimulerer epitelproliferasjon, noe som bidrar til den karakteristiske proliferative signaleringsevnen i neoplastiske celler ( 88 ). En annen mekanisme som spesifikke typer bakterier fremmer tumorutvikling er butyratproduserende bakterier, hvis overflod øker hos pasienter med tykktarmskreft (92).
Produksjon av metabolitten butyrat har komplekse fysiologiske effekter, inkludert induksjon av senescent epitel- og fibroblastceller. En musemodell av tykktarmskreft kolonisert med butyratproduserende bakterier utviklet flere svulster enn mus som mangler slike bakterier; Sammenhengen mellom butyratindusert senescens og økt tykktarmssvulstdannelse er påvist gjennom bruk av et senolytisk medikament som dreper senescerende celler, og svekker tumorvekst ( 92 ).
Videre har bakterielt produsert butyrat pleiotrope og paradoksale effekter på differensierte celler sammenlignet med udifferensierte (stam)celler i tykktarmsepitelet under tilstander hvor tarmbarrieren er forstyrret (dysbiose) og bakteriene er invasive, som påvirker for eksempel cellulær energi og metabolisme, histonprogresjons-immunisering, og (inntumorsykkel-immunisering) som immundemper adaptive immunresponser (93).
Faktisk involverer en bred handling av polymorfe mikrobiomer modulering av det adaptive og medfødte immunsystemet gjennom forskjellige veier, inkludert produksjon av "immunmodulerende" faktorer av bakterier som aktiverer skadesensorer på epitelceller eller fastboende immunceller, noe som fører til uttrykk for et mangfoldig repertoar av kjemokiner og cytokiner som kan forme immuncellene og epitoniske egenskaper. underliggende stroma og drenerende lymfeknuter.
I tillegg kan visse bakterier bryte både den beskyttende biofilmen og slimet i tykktarmsepitelet og forstyrre epitelcelle-celle-tette koblinger som til sammen opprettholder integriteten til den fysiske barrieren som normalt oppdeler tarmmikrobiomet. Ved invadering av stroma kan bakterier utløse både medfødte og adaptive immunresponser ved å fremkalle sekresjon av et repertoar av cytokiner og kjemokiner. En manifestasjon kan være dannelsen av tumorfremmende eller tumor-antagoniserende immunmikromiljøer, som følgelig beskytter mot eller letter tumorgenese og ondartet progresjon.
Følgelig kan moduleringen av de sammenvevde parametrene for (i) induksjon av (medfødt) tumorfremmende betennelse og (ii) flukt fra (adaptiv) immundestruksjon av karakteristiske mikrobiomer hos individuelle pasienter være assosiert ikke bare med prognose, men også med respons eller motstand mot immunterapier med immunkontrollpunkthemmere og andre terapeutiske manifestasjoner av tumorer og andre terapeutiske manifestasjoner. tumor-antagoniserende immunmikromiljøer, som følgelig oppstår Beskytte eller lette tumorutvikling og ondartet progresjon.
Følgelig kan moduleringen av de sammenvevde parametrene for (i) induksjon av (medfødt) tumorfremmende betennelse og (ii) flukt fra (adaptiv) immundestruksjon av karakteristiske mikrobiomer hos individuelle pasienter være assosiert ikke bare med prognose, men også med respons eller motstand mot immunterapier med immunkontrollpunkthemmere og andre terapeutiske manifestasjoner av tumorer og andre terapeutiske manifestasjoner. tumor-antagoniserende immunmikromiljøer, som følgelig oppstår beskytter eller letter tumorutvikling og ondartet progresjon).
Følgelig kan modulering av de sammenvevde parametrene for (i) induksjon av (medfødt) tumorfremmende betennelse og (ii) unnslippe fra (adaptiv) immunødeleggelse av særegne mikrobiomer hos individuelle pasienter være assosiert ikke bare med prognose, men også med respons eller motstand mot immunterapier med immunkontrollpunkthemmere og andre terapeutiske modaliteter (4–96, 96). Foreløpig proof-of-concept kommer fra nyere studier som viser gjenopprettet effekt av immunterapi etter transplantasjoner av fekal mikrobiota fra terapi-respondere til pasienter med melanom som hadde progrediert under tidligere behandling med immunkontrollpunktblokkade (97, 98).
De molekylære mekanismene som gjør at distinkte og variable komponenter i tarmmikrobiomet systemisk modulerer aktiviteten til det adaptive immunsystemet, forblir et vedvarende mysterium, enten ved å forsterke antitumorimmunresponser fremkalt av immunkontrollpunktblokkering eller snarere ved å indusere systemisk eller lokal (intratumoral) immunsuppresjon. En fersk studie har kastet lys: visse stammer av Enterococcus (og andre bakterier) uttrykker en peptidoglykanhydrolyase kalt SagA, som frigjør mukopeptider fra bakterieveggen, som deretter kan sirkulere systemisk og aktivere NOD2-mønsterreseptoren, som igjen øker T-celleresponsen og effektiviteten av sjekkpunktimmunterapi (99).
Andre immunregulerende molekyler produsert av spesifikke bakterielle underarter blir identifisert og funksjonelt vurdert, inkludert bakterieprodusert inosin, en hastighetsbegrensende metabolitt for T-celleaktivitet ( 100 ). Disse og andre eksempler begynner å avgrense de molekylære mekanismene som polymorfe mikrobiomer indirekte og systemisk modulerer tumorimmunobiologi, utover immunresponser som følger direkte fysiske interaksjoner mellom bakterier og immunsystemet (101, 102).
Bortsett fra årsakssammenhengene til tykktarmskreft og melanom, er den demonstrerte evnen til tarmmikrobiomet til å fremkalle ekspresjon av immunmodulerende kjemokiner og cytokiner som kommer inn i den systemiske sirkulasjonen, også tilsynelatende i stand til å påvirke kreftpatogenesen og respons på terapier i andre organer i kroppen (94, 95).
Et lysende eksempel gjelder utviklingen av kolangiokarsinomer i leveren: intestinal dysbiose tillater inngang og transport av bakterier og bakterielle produkter gjennom portvenen til leveren, der TLR4 uttrykt på hepatocytter utløses for å indusere ekspresjon av kjemokinet CXCL1, som rekrutterer CXSC-ekspressende celleekspressende celleceller (mye-celleekspressende) undertrykke naturlige drepeceller for å unngå immundestruksjon (103) og sannsynligvis formidle andre særegne evner (85). Som sådan er tarmmikrobiomet klart implisert som en muliggjørende funksjon som alternativt kan lette eller beskytte mot flere kreftformer.
Beyond the gut: Impliserer distinkte mikrobiomer i andre barrierevev
Nesten alle vev og organer som er direkte eller indirekte eksponert for det ytre miljø er også depoter for kommensale mikroorganismer ( 104 ). I motsetning til tarmen, hvor den symbiotiske rollen til mikrobiomet i metabolismen er godt anerkjent, dukker fortsatt de normale og patogene rollene til den bosatte mikrobiotaen på disse forskjellige stedene frem.
Det er tilsynelatende organ/vevsspesifikke forskjeller i konstitusjonen av de assosierte mikrobiomene i homeostase, aldring og kreft, med både overlappende og særegne arter og frekvenser til de i tykktarmen (104, 105). Videre gir assosiasjonsstudier økende bevis på at lokale tumorantagoniserende/beskyttende versus tumorfremmende vevsmikrobiomer, lik tarmmikrobiomet, kan modulere mottakelighet og patogenese for humane kreftformer som oppstår i deres assosierte organer (106 – 109).
Påvirkning av intratumoral mikrobiota?
Til slutt har patologer lenge erkjent at bakterier kan påvises i solide svulster, en observasjon som nå har blitt underbygget av sofistikerte profileringsteknologier. For eksempel, i en studie av 1526 svulster som spenner over syv menneskelige krefttyper (bein, hjerne, bryst, lunge, melanom, eggstokk og bukspyttkjertel), ble hver type karakterisert av et særegent mikrobiom, hovedsakelig lokalisert i kreftceller og immunceller. Innenfor hver tumortype har variasjoner i tumormikrobiomet blitt påvist og konkludert med å være assosiert med klinikopatologiske trekk (110).
Mikrobiota har på samme måte blitt påvist i de novo genetisk konstruerte musemodeller av lunge- og bukspyttkjertelkreft, og deres fravær i bakteriefrie mus og/eller deres opphevelse med antibiotika kan vise seg å svekke tumorigenese, og funksjonelt implisere tumormikrobiomet som en forløper til tumorfremmende betennelse og ondartet progresjon (11211, progresjon).
Assosiasjonsstudier i humant pankreas duktalt adenokarsinom og funksjonelle analyser via fekal transplantasjon i tumorbærende mus har vist at variasjoner i tumormikrobiomet – og det tilhørende tarmmikrobiomet – modulerer immunsystemets fenotyper og overlevelse ( 113 ). En viktig utfordring for fremtiden vil være å utvide disse implikasjonene til andre tumortyper og å skille de potensielt separerbare bidragene av konstitusjon og variasjon i tumormikrobiomet fra de fra tarmmikrobiomet (og det lokale opprinnelsesvevet), kanskje ved å identifisere spesifikke mikrobielle arter som er funksjonelt innflytelsesrike på ett eller annet sted.
Sammendrag
Spennende spørsmål for fremtiden inkluderer om mikrobiota som bor i forskjellige vev eller befolker begynnende neoplasmer har evnen til å bidra til eller forstyrre tilegnelsen av andre særegne evner utover immunmodulering og genomisk mutasjon, og dermed påvirke tumorutvikling og progresjon. Det er bevis på at visse bakteriearter direkte kan stimulere kjennetegnet ved proliferativ signalering, for eksempel i tykktarmsepitelet (88), og kan modulere vekstundertrykkelse ved å endre tumorsuppressoraktivitet i forskjellige deler av tarmen (114), mens direkte effekter på andre karakteristiske evner, som å unngå celledød, utløse, utløse, stimulerende angiogenese i forblir, samt unclear, angiogenese, generaliserbarhet av disse observasjonene til flere former for kreft hos mennesker.
Uansett er det stadig mer overbevisende argumenter for at polymorf variasjon i mikrobiomene i tarmen og andre organer representerer et særegent aktiverende trekk for tilegnelse av særegne ferdigheter (fig. 4), selv om det overlapper med og komplementerer de for genomets ustabilitet og mutasjon, og svulstfremmende betennelse.
Aldrende celler
Cellulær senescens er en typisk irreversibel form for proliferativ arrestasjon som sannsynligvis utviklet seg som en beskyttende mekanisme for å opprettholde vevshomeostase, tilsynelatende som en komplementær mekanisme til programmert celledød som tjener til å inaktivere og, etter hvert, fjerne syke, dysfunksjonelle eller på annen måte unødvendige celler. I tillegg til å stenge av celledelingssyklusen, produserer senescensprogrammet endringer i cellemorfologi og metabolisme og, mest dyptgripende, aktivering av en senescensassosiert sekretorisk fenotype (SASP), som involverer frigjøring av en mengde bioaktive proteiner, inkludert kjemokiner.
Cytokiner og proteaser, hvis identitet avhenger av celle- og vevstypen som en senescent celle oppstår fra ( 115–117). Aldring kan induseres i celler av en rekke forhold, inkludert mikromiljøbelastninger som næringssult og DNA-skader, samt skade på organeller og cellulær infrastruktur og ubalanser i cellulære signalnettverk (115, 117), som alle har skjedd i sammenheng med den observerte økningen i frekvensen av senescent celle i ulike organeller (8) 119).
Cellulær senescens har lenge vært ansett som en beskyttende mekanisme mot neoplasi, som gjør at kreftceller gjennomgår senescens ( 120 ). De fleste av de ovennevnte initiativtakerne til senescensprogrammet er assosiert med malignitet, spesielt DNA-skader som følge av avvikende hyperproliferasjon, såkalt onkogenindusert senescens på grunn av hyperaktivert signalering og terapiindusert senescens som følge av cellulær og genomisk skade forårsaket av kjemoterapi og strålebehandling.
Det er faktisk veletablerte eksempler på de beskyttende fordelene ved senescens for å begrense malign progresjon (118, 119). Tvert imot viser imidlertid en voksende mengde bevis akkurat det motsatte: I visse sammenhenger stimulerer senescentceller differensielt tumorutvikling og ondartet progresjon (119, 121).
I en innsiktsfull casestudie ble senescerende celler i aldrende mus farmakologisk ablaterte, og spesifikt utarmer senescentceller som karakteristisk uttrykker cellesyklushemmeren p16 – INK4a: i tillegg til å forsinke flere aldersrelaterte symptomer, resulterte dette i uttømming av senescentceller i aldrende mus med redusert incidens2-kreftdød (1).
Hovedmekanismen som senescentceller fremmer tumorfenotyper med antas å være SASP, som har vist seg å være i stand til å formidle signalmolekyler (og proteaser som aktiverer og/eller deaktiverer) på en parakrin måte for å formidle typiske evner. I ulike eksperimentelle systemer har senescent kreftceller således vist seg å bidra på ulike måter til proliferativ signalering, unngå apoptose, indusere angiogenese, stimulere invasjon og metastaser og undertrykke tumorimmunitet (116, 118, 120, 121).
Enda en fasett av effektene av senescent kreftceller på kreftfenotyper involverer forbigående, reversible senescent celletilstander, der senescerende kreftceller kan unnslippe sin SASP-uttrykkende, ikke-proliferative tilstand og gjenoppta celleproliferasjon og manifestasjon av de tilhørende egenskapene til en fullt levedyktig onkogencelle (44).
Slik forbigående senescens er best dokumentert i tilfeller av terapiresistens (44), som representerer en form for hvile som unngår terapeutisk målretting av prolifererende kreftceller, men som kan vise seg mer bredt effektivt i andre stadier av tumorutvikling, ondartet progresjon og metastasering.
Videre er de kjennetegnsfremmende evnene til senescent celler ikke begrenset til senescent kreftceller. Kreftassosierte fibroblaster (CAF) har vist seg å senescere i svulster, noe som gir opphav til senescerende CAF som har vist seg å være tumorfremmende ved å gi karakteristiske evner til kreftceller i TME (115, 116, 121).
Videre er eldre fibroblaster i normalt vev, delvis dannet av naturlig aldring eller miljøfornærmelser, på samme måte involvert i remodellering av vevsmikromiljøer via deres SASP for å gi parakrin støtte for lokal invasjon (såkalte "felteffekter") og fjernmetastaser (116) av neoplasmer som utvikler seg i nærheten.
Videre har aldrende fibroblaster i aldrende hud vist seg å rekruttere – via deres SASP – medfødte immunceller som både er immunsuppressive overfor adaptive antitumorimmunresponser forankret av CD8 T-celler og stimulerer hudtumorvekst (123), sistnevnte effekt gjenspeiler muligens parakrine bidrag fra slike medfødte immunceller og nøytrofile celler (myeloidceller) reflekterer.
Selv om det er mindre godt etablert, virker det sannsynlig at andre rikelige stromale celler som befolker spesifikke tumormikromiljøer vil gjennomgå senescens, og derved modulere kreftkarakteristikker og resulterende tumorfenotyper. For eksempel kan terapi-induserte senescent tumorendotelceller forsterke proliferasjon, invasjon og metastase i brystkreftmodeller (124, 125).
Slike bevis garanterer absolutt undersøkelse i andre tumortyper for å evaluere generell senescens av fibroblaster, endotelceller og andre stromale celler som en drivkraft i tumorutvikling. Også foreløpig uklare er de regulatoriske mekanismene og funksjonelle determinantene som gjør at en bestemt senescent celletype i en bestemt TME fremkaller en tumor-fremmende versus en tumor-antagoniserende SASP, som tilsynelatende kan induseres alternativt i samme senescent celletype, kanskje av forskjellige initiatorer når nedsenket i karakteristiske fysiologiske og neoplastiske mikromiljøer.
Sammendrag
Konseptet om at svulster består av genetisk transformerte kreftceller som interagerer med og drar nytte av rekrutterte og epigenetisk/fenotypisk korrupte hjelpeceller (stromale) celler har blitt etablert som avgjørende for patogenesen av kreft. Betraktningene diskutert ovenfor og beskrevet i oversiktene og rapportene som er sitert her (og andre steder) argumenterer overbevisende for at senescentceller (uavhengig av cellulær opprinnelse) bør vurderes for inkludering i listen over funksjonelt signifikante celler i tumormikromiljøet (fig. 5). Derfor bør senescent celler vurderes i søket etter inngående kunnskap om kreftmekanismer. Videre motiverer anerkjennelsen av deres betydning det sekundære målet om å terapeutisk målrette tumorfremmende senescentceller av alle konstitusjoner, det være seg gjennom farmakologisk eller immunologisk ablasjon eller ved å omprogrammere SASP til tumorantagoniserende varianter (115, 121, 126).
Figur 5

Heterogene kreftcelleundertyper og stromacelletyper og -undertyper er funksjonelt integrert i manifestasjonene av svulster som ulovlige organer. Økende bevis tyder på at senescentcellederivater av mange av disse cellulære komponentene i TME og deres variable SASP-er er involvert i moduleringen av kjennetegnsevner og resulterende tumorfenotyper. Varemerkene til kreftgrafikken ble adoptert fra Hanahan og Weinberg (2).
Avsluttende bemerkninger
Mens de åtte kjennetegnene til kreft og deres to støttende trekk har vist seg å ha varig heuristisk verdi i konseptualiseringen av kreft, antyder betraktningene som er presentert ovenfor at det kan være nye fasetter av en viss generalitet og derfor viktighet for en mer fullstendig forståelse av kompleksiteten, mekanismene og manifestasjonene av sykdommen. Ved å anvende metrikken for merkbar, om ikke fullstendig, uavhengighet fra de 10 kjerneattributtene, kan det argumenteres for at disse fire parametrene – ved ytterligere validering og generalisering utover de presenterte casestudiene – godt kan integreres i kjennetegnene til kreftskjemaet (fig. 6).
Derfor kan cellulær plastisitet legges til listen over fremtredende egenskaper. Mens den åttende kjernen og denne nouveau-evnen hver konseptuelt kan skilles ut ved sin definisjon som kjennetegn, er aspekter av deres regulering i det minste delvis knyttet til noen og kanskje mange kreftformer. For eksempel er flere kjennetegn koordinert modulert av kanoniske onkogene drivere i noen tumortyper, inkludert
- (I) KRAS ( https://cancer.sanger.ac.uk/cosmic/census-page/KRAS ),
- (II) MYC ( https://cancer.sanger.ac.uk/cosmic/census-page/MYC ),
- (III) NOTCH ( https://cancer.sanger.ac.uk/cosmic/census-page/NOTCH1 ; Ref. 127) und
- (IV) TP53 ( https://cancer.sanger.ac.uk/cosmic/census-page/TP53 )
Figur 6
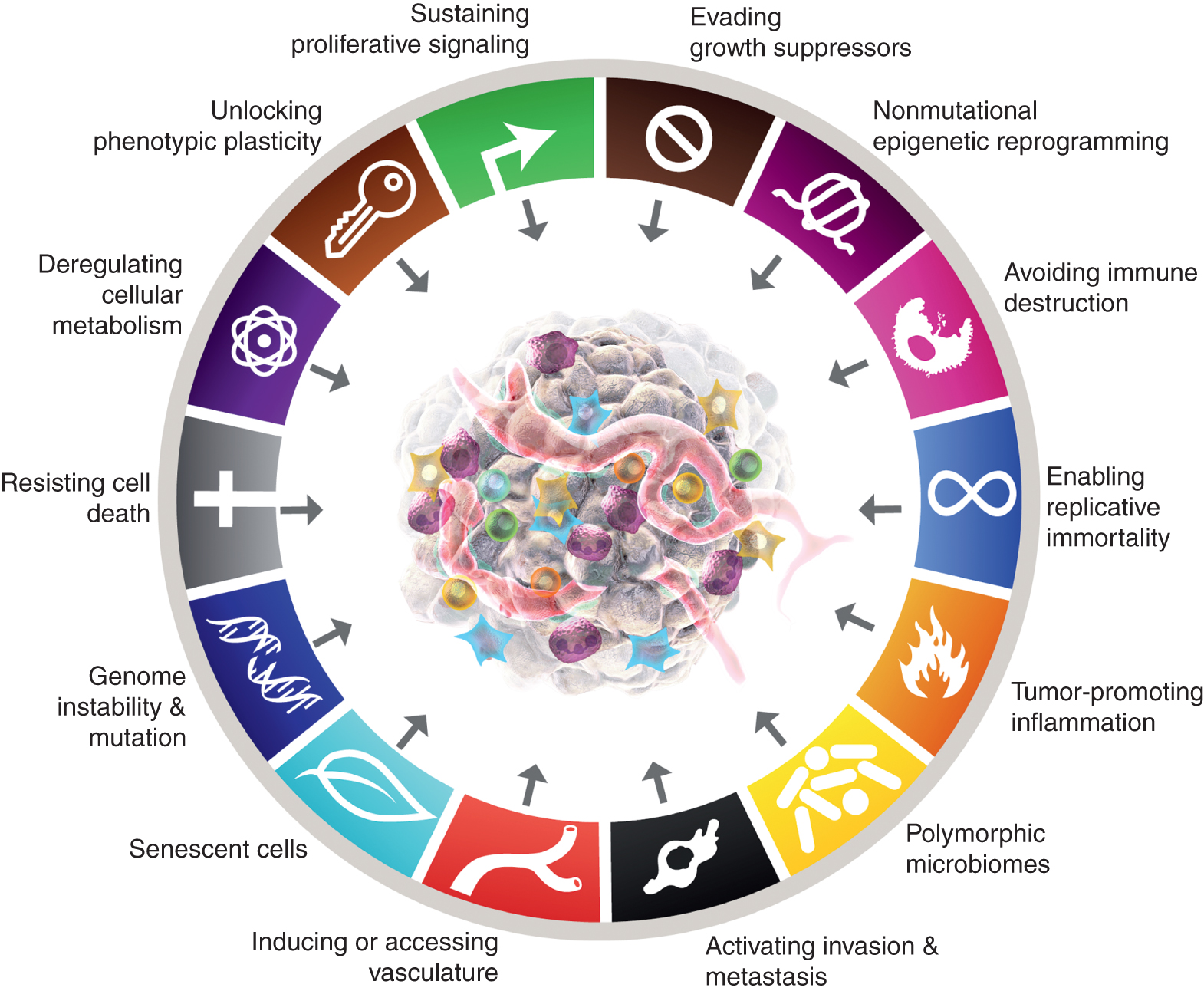
De kanoniske og forventede nye tilleggene til «Kreftens kjennetegn» vises. Denne artikkelen tar opp muligheten, med sikte på å stimulere debatt, diskusjon og eksperimentell utdyping, at noen eller alle de fire nye parameterne vil bli anerkjent som generiske for flere former for kreft hos mennesker og derfor egnet for integrering i kjernekonseptualiseringen av kreftens kjennetegn. Varemerkene til kreftgrafikken ble adoptert fra Hanahan og Weinberg (2).
I tillegg til å legge til cellulær plastisitet til listen, kan ikke-mutasjonsfremmende epigenetisk omprogrammering og polymorfe variasjoner integreres i organ/vevsmikrobiomer som mekanistiske determinanter – muliggjørende egenskaper – gjennom hvilke særegne evner erverves, sammen med svulstfremmende betennelse (selv delvis sammenkoblet med mikrobiomet), utover de ovenfor nevnte mutasjonene og andre kogener.
Til slutt kan senescerende celler med forskjellig opprinnelse - inkludert kreftceller og forskjellige stromaceller - som funksjonelt bidrar til utvikling og ondartet progresjon av kreft, om enn på markant forskjellige måter fra de til deres ikke-senescente brødre, inkluderes som generiske komponenter av TME. Oppsummert er det tenkt at utplasseringen av disse foreløpige "eksperimentelle ballongene" vil stimulere til debatt, diskusjon og ytterligere eksperimentell undersøkelse i kreftforskningsmiljøet om de definerende konseptuelle parameterne for kreftbiologi, genetikk og patogenese.
Referanser
- Hanahan D , Weinberg RA . The hallmarks of cancer. Cell 2000;100:57–70.
- Hanahan D , Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Yuan S , Norgard RJ , Stanger BZ . Cellular plasticity in cancer. Cancer Discov 2019;9:837–51.
- Barker N , Ridgway RA , van Es JH , van de Wetering M , Begthel H , van den Born M et al . Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608–11.
- Perekatt AO , Shah PP , Cheung S , Jariwala N , Wu A , Gandhi V et al . SMAD4 suppresses WNT-driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res 2018;78:4878–90.
- Shih IM , Wang TL , Traverso G , Romans K , Hamilton SR , Ben-Sasson S et al . Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A 2001;98:2640–5.
- Ordóñez-Morán P , Dafflon C , Imajo M , Nishida E , Huelsken J . HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 2015;28:815–29.
- Tan SH , Barker N . Stemming colorectal cancer growth and metastasis: HOXA5 forces cancer stem cells to differentiate. Cancer Cell 2015;28:683–5.
- Köhler C , Nittner D , Rambow F , Radaelli E , Stanchi F , Vandamme N et al . Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 2017;21:679–93.
- Shah M , Bhoumik A , Goel V , Dewing A , Breitwieser W , Kluger H et al . A role for ATF2 in regulating MITF and melanoma development. PLoS Genet 2010;6:e1001258.
- Claps G , Cheli Y , Zhang T , Scortegagna M , Lau E , Kim H et al . A transcriptionally inactive ATF2 variant drives melanomagenesis. Cell Rep 2016;15:1884–92.
- Saghafinia S , Homicsko K , Di Domenico A , Wullschleger S , Perren A , Marinoni I et al . Cancer cells retrace a stepwise differentiation program during malignant progression. Cancer Discov 2021;11:2638–57.
- Yu X-X , Qiu W-L , Yang L , Zhang Y , He M-Y , Li L-C et al . Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J 2019;38:e100164.
- de Thé H . Differentiation therapy revisited. Nat Rev Cancer 2018;18:117–27.
- He LZ , Merghoub T , Pandolfi PP . In vivo analysis of the molecular pathogenesis of acute promyelocytic leukemia in the mouse and its therapeutic implications. Oncogene 1999;18:5278–92.
- Warrell RP , de Thé H , Wang ZY , Degos L . Acute promyelocytic leukemia. N Engl J Med 1993;329:177–89.
- Bots M , Verbrugge I , Martin BP , Salmon JM , Ghisi M , Baker A et al . Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014;123:1341–52.
- Ferrara FF , Fazi F , Bianchini A , Padula F , Gelmetti V , Minucci S et al . Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res 2001;61:2–7.
- Kaufman CK , Mosimann C , Fan ZP , Yang S , Thomas AJ , Ablain J et al . A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016;351:aad2197.
- Morris JP , Yashinskie JJ , Koche R , Chandwani R , Tian S , Chen C-C et al . α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019;573:595–9.
- Saha SK , Parachoniak CA , Ghanta KS , Fitamant J , Ross KN , Najem MS et al . Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110–4.
- Dang L , Su S-SM . Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem 2017;86:305–31.
- Waitkus MS , Diplas BH , Yan H . Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018;34:186–95.
- Phan TG , Croucher PI . The dormant cancer cell life cycle. Nat Rev Cancer 2020;20:398–411.
- Jiang M , Azevedo-Pouly AC , Deering TG , Hoang CQ , DiRenzo D , Hess DA et al . MIST1 and PTF1 collaborate in feed-forward regulatory loops that maintain the pancreatic acinar phenotype in adult mice. Mol Cell Biol 2016;36:2945–55.
- Krah NM , Narayanan SM , Yugawa DE , Straley JA , Wright CVE , MacDonald RJ et al . Prevention and reversion of pancreatic tumorigenesis through a differentiation-based mechanism. Dev Cell 2019;50:744–54.
- Krah NM , De La O J-P , Swift GH , Hoang CQ , Willet SG , Chen Pan F et al . The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015;4:e07125.
- Shi G , DiRenzo D , Qu C , Barney D , Miley D , Konieczny SF . Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 2013;32:1950–8.
- Kopp JL , von Figura G , Mayes E , Liu F-F , Dubois CL , Morris JP et al . Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50.
- Julian LM , McDonald AC , Stanford WL . Direct reprogramming with SOX factors: masters of cell fate. Curr Opin Genet Dev 2017;46:24–36.
- Grimm D , Bauer J , Wise P , Krüger M , Simonsen U , Wehland M et al . The role of SOX family members in solid tumours and metastasis. Semin Cancer Biol 2020;67:122–53.
- Mu P , Zhang Z , Benelli M , Karthaus WR , Hoover E , Chen C-C et al . SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84–8.
- Von Hoff DD , LoRusso PM , Rudin CM , Reddy JC , Yauch RL , Tibes R et al . Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72.
- Biehs B , Dijkgraaf GJP , Piskol R , Alicke B , Boumahdi S , Peale F et al . A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018;562:429–33.
- Boumahdi S , de Sauvage FJ . The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19:39–56.
- Groves SM , Ireland A , Liu Q , Simmons AJ , Lau K , Iams WT et al . Cancer Hallmarks Define a Continuum of Plastic Cell States between Small Cell Lung Cancer Archetypes [Internet]. Systems Biology; 2021 Jan. Available from: http://biorxiv.org/lookup/doi/10.1101/2021.01.22.427865.
- LaFave LM , Kartha VK , Ma S , Meli K , Del Priore I , Lareau C et al . Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 2020;38:212–28.
- Marjanovic ND , Hofree M , Chan JE , Canner D , Wu K , Trakala M et al . Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 2020;38:229–46.
- Drapkin BJ , Minna JD . Studying lineage plasticity one cell at a time. Cancer Cell 2020;38:150–2.
- Inoue Y , Nikolic A , Farnsworth D , Liu A , Ladanyi M , Somwar R et al . Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer [Internet]. Cancer Biology; 2020 Nov. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.11.12.368522.
- Dravis C , Chung C-Y , Lytle NK , Herrera-Valdez J , Luna G , Trejo CL et al . Epigenetic and transcriptomic profiling of mammary gland development and tumor models disclose regulators of cell state plasticity. Cancer Cell 2018;34:466–82.
- Malta TM , Sokolov A , Gentles AJ , Burzykowski T , Poisson L , Weinstein JN et al . Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–54.
- Miao Z-F , Lewis MA , Cho CJ , Adkins-Threats M , Park D , Brown JW et al . A dedicated evolutionarily conserved molecular network licenses differentiated cells to return to the cell cycle. Dev Cell 2020;55:178–94.
- De Blander H , Morel A-P , Senaratne AP , Ouzounova M , Puisieux A . Cellular plasticity: a route to senescence exit and tumorigenesis. Cancers 2021;13:4561.
- Merrell AJ , Stanger BZ . Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 2016;17:413–25.
- Baylin SB , Jones PA . Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 2016;8:a019505.
- Flavahan WA , Gaskell E , Bernstein BE . Epigenetic plasticity and the hallmarks of cancer. Science 2017;357:eaal2380.
- Jones PA , Issa J-PJ , Baylin S . Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630–41.
- Huang S . Tumor progression: Chance and necessity in Darwinian and Lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol 2012;110:69–86.
- Darwiche N . Epigenetic mechanisms and the hallmarks of cancer: an intimate affair. Am J Cancer Res 2020;10:1954–78.
- Feng Y , Liu X , Pauklin S . 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell 2021;12:440–54.
- Nam AS , Chaligne R , Landau DA . Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet 2021;22:3–18.
- Bitman-Lotan E , Orian A . Nuclear organization and regulation of the differentiated state. Cell Mol Life Sci CMLS 2021;78:3141–58.
- Goldberg AD , Allis CD , Bernstein E . Epigenetics: a landscape takes shape. Cell 2007;128:635–8.
- Zeng Y , Chen T . DNA methylation reprogramming during mammalian development. Genes 2019;10:257.
- Hegde AN , Smith SG . Recent developments in transcriptional and translational regulation underlying long-term synaptic plasticity and memory. Learn Mem 2019;26:307–17.
- Kim S , Kaang B-K . Epigenetic regulation and chromatin remodeling in learning and memory. Exp Mol Med 2017;49:e281.
- Thienpont B , Van Dyck L , Lambrechts D . Tumors smother their epigenome. Mol Cell Oncol 2016;3:e1240549.
- Gameiro PA , Struhl K . Nutrient deprivation elicits a transcriptional and translational inflammatory response coupled to decreased protein synthesis. Cell Rep 2018;24:1415–24.
- Lin GL , Monje M . Understanding the deadly silence of posterior fossa A ependymoma. Mol Cell 2020;78:999–1001.
- Michealraj KA , Kumar SA , Kim LJY , Cavalli FMG , Przelicki D , Wojcik JB et al . Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell 2020;181:1329–45.
- Bakir B , Chiarella AM , Pitarresi JR , Rustgi AK . EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol 2020;30:764–76.
- Gupta PB , Pastushenko I , Skibinski A , Blanpain C , Kuperwasser C . Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 2019;24:65–78.
- Lambert AW , Weinberg RA . Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer 2021;21:325–38.
- Lindner P , Paul S , Eckstein M , Hampel C , Muenzner JK , Erlenbach-Wuensch K et al . EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis 2020;11:147.
- Javaid S , Zhang J , Anderssen E , Black JC , Wittner BS , Tajima K et al . Dynamic chromatin modification sustains epithelial-mesenchymal transition following inducible expression of Snail-1. Cell Rep 2013;5:1679–89.
- Serrano-Gomez SJ , Maziveyi M , Alahari SK . Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 2016;15:18.
- Skrypek N , Goossens S , De Smedt E , Vandamme N , Berx G . Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet TIG 2017;33:943–59.
- Li L , Hanahan D . Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 2013;153:86–100.
- Li L , Zeng Q , Bhutkar A , Galván JA , Karamitopoulou E , Noordermeer D et al . GKAP acts as a genetic modulator of NMDAR signaling to govern invasive tumor growth. Cancer Cell 2018;33:736–51.
- Mohammadi H , Sahai E . Mechanisms and impact of altered tumour mechanics. Nat Cell Biol 2018;20:766–74.
- Odenthal J , Takes R , Friedl P . Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis 2016;37:1117–28.
- Torres CM , Biran A , Burney MJ , Patel H , Henser-Brownhill T , Cohen A-HS et al . The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 2016;353:aaf1644.
- Puram SV , Tirosh I , Parikh AS , Patel AP , Yizhak K , Gillespie S et al . Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171:1611–24.
- Kinker GS , Greenwald AC , Tal R , Orlova Z , Cuoco MS , McFarland JM et al . Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 2020;52:1208–18.
- Murtha M , Esteller M . Extraordinary cancer epigenomics: thinking outside the classical coding and promoter box. Trends Cancer 2016;2:572–84.
- Nebbioso A , Tambaro FP , Dell’Aversana C , Altucci L . Cancer epigenetics: moving forward. PLoS Genet 2018;14:e1007362.
- Tavernari D , Battistello E , Dheilly E , Petruzzella AS , Mina M , Sordet-Dessimoz J et al . Non-genetic evolution drives lung adenocarcinoma spatial heterogeneity and progression. Cancer Discov 2021;11:1490–507.
- Heyn H , Vidal E , Ferreira HJ , Vizoso M , Sayols S , Gomez A et al . Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 2016;17:11.
- Saghafinia S , Mina M , Riggi N , Hanahan D , Ciriello G . Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 2018;25:1066–80.
- Audia JE , Campbell RM . Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8:a019521.
- Corces MR , Granja JM , Shams S , Louie BH , Seoane JA , Zhou W et al . The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898.
- Esteve-Puig R , Bueno-Costa A , Esteller M . Writers, readers and erasers of RNA modifications in cancer. Cancer Lett 2020;474:127–37.
- Janin M , Coll-SanMartin L , Esteller M . Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer 2020;19:70.
- Hanahan D , Coussens LM . Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–22.
- Lu Z , Zou J , Li S , Topper MJ , Tao Y , Zhang H et al . Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020;579:284–90.
- Thomas S , Izard J , Walsh E , Batich K , Chongsathidkiet P , Clarke G et al . The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res 2017;77:1783–812.
- Dzutsev A , Badger JH , Perez-Chanona E , Roy S , Salcedo R , Smith CK et al . Microbes and cancer. Annu Rev Immunol 2017;35:199–228.
- Helmink BA , Khan MAW , Hermann A , Gopalakrishnan V , Wargo JA . The microbiome, cancer, and cancer therapy. Nat Med 2019;25:377–88.
- Sears CL , Garrett WS . Microbes, microbiota, and colon cancer. Cell Host Microbe 2014;15:317–28.
- Pleguezuelos-Manzano C , Puschhof J , Rosendahl Huber A , van Hoeck A , Wood HM , Nomburg J et al . Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020;580:269–73.
- Okumura S , Konishi Y , Narukawa M , Sugiura Y , Yoshimoto S , Arai Y et al . Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat Commun 2021;12:5674.
- Salvi PS , Cowles RA . Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in homeostasis and disease. Cells 2021;10:1775.
- Fessler J , Matson V , Gajewski TF . Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 2019;7:108.
- Gopalakrishnan V , Helmink BA , Spencer CN , Reuben A , Wargo JA . The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 2018;33:570–80.
- Zitvogel L , Ma Y , Raoult D , Kroemer G , Gajewski TF . The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–70.
- Baruch EN , Youngster I , Ben-Betzalel G , Ortenberg R , Lahat A , Katz L et al . Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021;371:602–9.
- Davar D , Dzutsev AK , McCulloch JA , Rodrigues RR , Chauvin J-M , Morrison RM et al . Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021;371:595–602.
- Griffin ME , Espinosa J , Becker JL , Luo J-D , Carroll TS , Jha JK et al . Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 2021;373:1040–6.
- Mager LF , Burkhard R , Pett N , Cooke NCA , Brown K , Ramay H et al . Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369:1481–9.
- Ansaldo E , Belkaid Y . How microbiota improve immunotherapy. Science 2021;373:966–7.
- Ansaldo E , Farley TK , Belkaid Y . Control of immunity by the microbiota. Annu Rev Immunol 2021;39:449–79.
- Zhang Q , Ma C , Duan Y , Heinrich B , Rosato U , Diggs LP et al . Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov 2021;11:1248–67.
- Ding T , Schloss PD . Dynamics and associations of microbial community types across the human body. Nature 2014;509:357–60.
- Byrd AL , Liu M , Fujimura KE , Lyalina S , Nagarkar DR , Charbit B et al . Gut microbiome stability and dynamics in healthy donors and patients with non-gastrointestinal cancers. J Exp Med 2021;218:e20200606.
- Healy CM , Moran GP . The microbiome and oral cancer: more questions than answers. Oral Oncol 2019;89:30-3.
- Swaney MH , Kalan LR . Living in your skin: microbes, molecules and mechanisms. Infect Immun 2021;89:e00695–20.
- Willis JR , Gabaldón T . The human oral microbiome in health and disease: from sequences to ecosystems. Microorganisms 2020;8:308.
- Xu J , Peng J-J , Yang W , Fu K , Zhang Y . Vaginal microbiomes and ovarian cancer: a review. Am J Cancer Res 2020;10:743–56.
- Nejman D , Livyatan I , Fuks G , Gavert N , Zwang Y , Geller LT et al . The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80.
- Jin C , Lagoudas GK , Zhao C , Bullman S , Bhutkar A , Hu B et al . Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019;176:998–1013.
- Pushalkar S , Hundeyin M , Daley D , Zambirinis CP , Kurz E , Mishra A et al . The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 2018;8:403–16.
- McAllister F , Khan MAW , Helmink B , Wargo JA . The tumor microbiome in pancreatic cancer: bacteria and beyond. Cancer Cell 2019;36:577–9.
- Kadosh E , Snir-Alkalay I , Venkatachalam A , May S , Lasry A , Elyada E et al . The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020;586:133–8.
- Birch J , Gil J . Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34:1565–76.
- Faget DV , Ren Q , Stewart SA . Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 2019;19:439–53.
- Gorgoulis V , Adams PD , Alimonti A , Bennett DC , Bischof O , Bishop C et al . Cellular senescence: defining a path forward. Cell 2019;179:813–27.
- He S , Sharpless NE . Senescence in health and disease. Cell 2017;169:1000–11.
- Kowald A , Passos JF , Kirkwood TBL . On the evolution of cellular senescence. Aging Cell 2020;19:e13270.
- Lee S , Schmitt CA . The dynamic nature of senescence in cancer. Nat Cell Biol 2019;21:94–101.
- Wang B , Kohli J , Demaria M . Senescent cells in cancer therapy: friends or foes? Trends Cancer 2020;6:838–57.
- Baker DJ , Childs BG , Durik M , Wijers ME , Sieben CJ , Zhong J et al . Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–9.
- Ruhland MK , Loza AJ , Capietto A-H , Luo X , Knolhoff BL , Flanagan KC et al . Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 2016;7:11762.
- Hwang HJ , Lee Y-R , Kang D , Lee HC , Seo HR , Ryu J-K et al . Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett 2020;490:100–10.
- Wang D , Xiao F , Feng Z , Li M , Kong L , Huang L et al . Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res 2020;22:103.
- Amor C , Feucht J , Leibold J , Ho Y-J , Zhu C , Alonso-Curbelo D et al . Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020;583:127–32.
- Aster JC , Pear WS , Blacklow SC . The varied roles of notch in cancer. Annu Rev Pathol 2017;12:245–75.
- Kuczynski EA , Vermeulen PB , Pezzella F , Kerbel RS , Reynolds AR . Vessel co-option in cancer. Nat Rev Clin Oncol 2019;16:469–93.
Google ScholarCrossref