 Suche
Suche
 Mein Konto
Mein Konto
Značilnosti raka: nove razsežnosti

Predgovor Značilnosti konceptualizacije raka so hevristično orodje za destilacijo ogromne kompleksnosti fenotipov in genotipov raka v predhodni nabor osnovnih načel. Ko je znanje o mehanizmih raka napredovalo, so se drugi vidiki bolezni pojavili kot možne izboljšave. To vzbuja možnost, da sta fenotipska plastičnost in neurejena diferenciacija izrazita značilna sposobnost in da nemutacijsko epigenetsko reprogramiranje in polimorfni mikrobiomi predstavljata značilne omogočitvene lastnosti, ki olajšajo pridobivanje značilnih sposobnosti. Poleg tega lahko stare celice različnega izvora dodamo na seznam funkcionalno pomembnih tipov celic v tumorskem mikrookolju. Pomeni, da je rak strašljiv v...

Značilnosti raka: nove razsežnosti
Predgovor
Značilnosti konceptualizacije raka so hevristično orodje za destilacijo ogromne kompleksnosti fenotipov in genotipov raka v predhodni nabor osnovnih načel. Ko je znanje o mehanizmih raka napredovalo, so se drugi vidiki bolezni pojavili kot možne izboljšave. To vzbuja možnost, da sta fenotipska plastičnost in neurejena diferenciacija izrazita značilna sposobnost in da nemutacijsko epigenetsko reprogramiranje in polimorfni mikrobiomi predstavljata značilne omogočitvene lastnosti, ki olajšajo pridobivanje značilnih sposobnosti. Poleg tega lahko stare celice različnega izvora dodamo na seznam funkcionalno pomembnih tipov celic v tumorskem mikrookolju.
Pomen
Rak je zastrašujoč zaradi širine in obsega svoje raznolikosti, ki vključuje genetiko, biologijo celic in tkiv, patologijo in odziv na terapijo. Vse močnejša eksperimentalna in računalniška orodja in tehnologije zagotavljajo plaz »velikih podatkov« o neštetih manifestacijah bolezni, ki jih zajema rak. Integrativni koncept, utelešen v značilnostih raka, pomaga destilirati to kompleksnost v vse bolj logično znanost, predhodne nove razsežnosti, predstavljene v tej perspektivi, pa lahko dodajo vrednost temu prizadevanju za boljše razumevanje mehanizmov karcinogeneze in malignega napredovanja ter uporabo tega znanja v medicini raka.
uvod
Znaki raka so bili predlagani kot nabor funkcionalnih sposobnosti, ki jih pridobijo človeške celice, ko se premaknejo iz normalnega stanja v neoplastično rastno stanje, natančneje zmožnosti, ki so ključne za njihovo sposobnost tvorbe malignih tumorjev. V teh člankih ( 1, 2 ), z Bobom Weinbergom sva naštela, kar sva si zamislila kot skupne značilnosti, ki združujejo vse vrste rakavih celic na ravni celičnega fenotipa. Namen je bil zagotoviti konceptualni okvir, ki bi omogočil racionalizacijo kompleksnih fenotipov različnih tipov in variant človeških tumorjev v povezavi s skupnim nizom osnovnih celičnih parametrov. Sprva smo predvideli komplementarno vključitev šestih različnih zmogljivosti blagovnih znamk, kasneje pa smo to število razširili na osem.
Na to formulacijo je vplivalo spoznanje, da se človeški raki razvijejo kot produkt večstopenjskih procesov in da bi lahko pridobitev teh funkcionalnih sposobnosti na nek način pripisali ločenim stopnjam tumorske patogeneze. Raznolikost maligne patogeneze, ki zajema več vrst tumorjev in naraščajočo množico podtipov, vključuje različne aberacije (in s tem pridobljene sposobnosti in lastnosti), ki so posledica tkivno specifičnih ovir, ki jih je med določenimi potemi tumorigeneze nujno zaobiti. Čeprav se zavedamo, da so lahko takšni specializirani mehanizmi koristni, smo določitev znakov omejili na parametre, ki imajo širok vpliv na spekter človeških rakov.
Osem značilnosti trenutno vključuje (slika 1, levo) pridobljene sposobnosti za vzdrževanje proliferativne signalizacije, izogibanje zaviralcem rasti, upiranje celični smrti, omogočanje replikativne nesmrtnosti, induciranje/dostop do žil, aktiviranje invazije in metastaz, reprogramiranje celičnega metabolizma in izogibanje uničenju imunskega sistema. V najnovejši elaboraciji tega koncepta (2) sta bila deregulacija celičnega metabolizma in izogibanje uničenju imunskega sistema razmejena kot "nastajajoča znamenja", zdaj, enajst let kasneje, pa je očitno, da se lahko podobno kot prvotnih šest obravnavata kot temeljna znaka raka in sta kot taka vključena v trenutno pripoved (slika 1, levo).
Slika 1
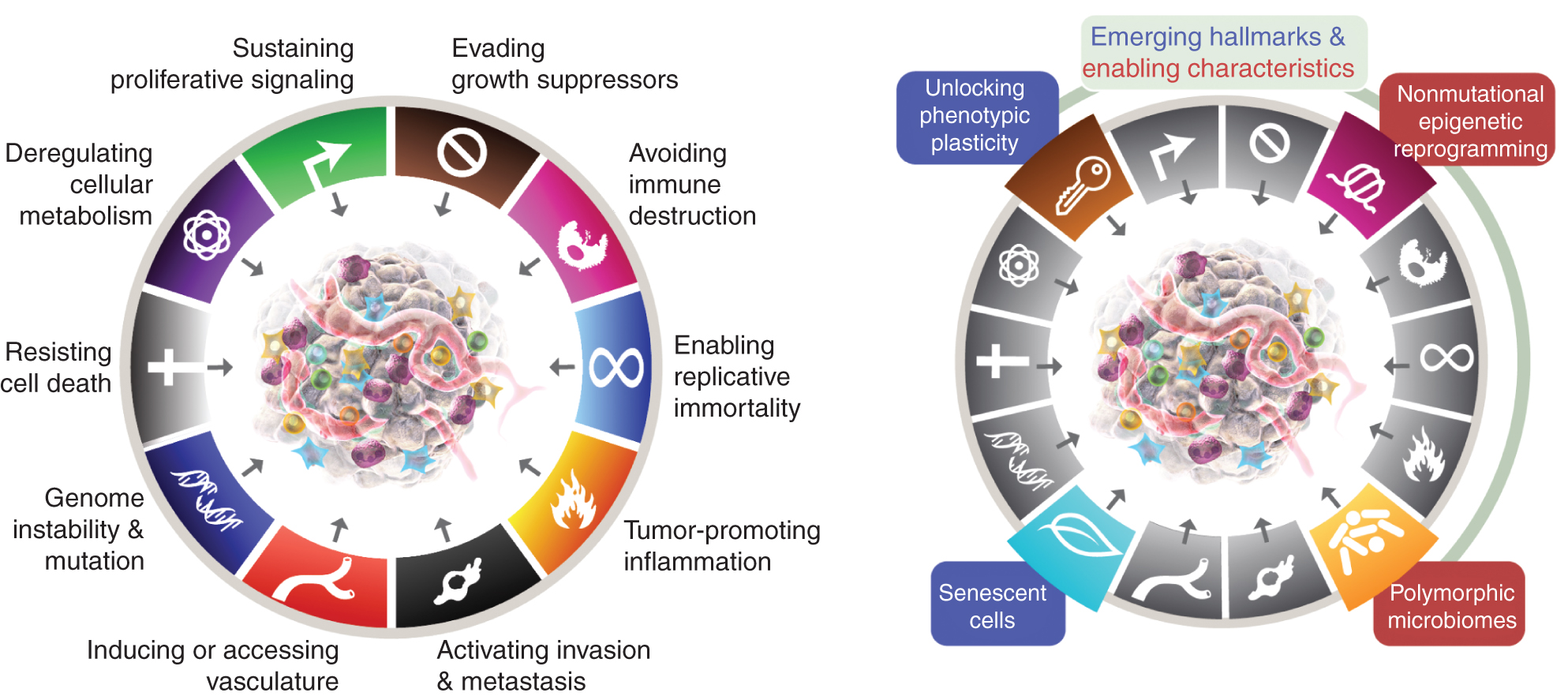
Značilnosti raka trenutno utelešajo osem značilnih sposobnosti in dve podporni lastnosti. Poleg šestih pridobljenih zmožnosti - znakov raka - predlaganih leta 2000 (1), sta bili dve predhodni "nastali značilnosti", uvedeni leta 2011 (2) - celična energetika (zdaj pogosteje imenovana kot "reprogramiranje celičnega metabolizma") in "izogibanje imunskemu uničenju" - dovolj potrjeni, da se štejeta za del osrednjega sklopa.
Glede na naraščajoče priznanje, da je tumorje mogoče ustrezno vaskularizirati, bodisi s preklopom na angiogenezo ali s sodelovanjem normalne tkivne vaskulature (128), je ta značilnost tudi širše opredeljena kot sposobnost induciranja ali drugačnega dostopa do vaskulature, ki podpira rast tumorja predvsem z invazijo in metastazami.
Nadaljevanje iz leta 2011 je vključevalo tudi »vnetje, ki spodbuja tumor« kot drugo omogočitveno lastnost, ki je dopolnjevala vsesplošno »nestabilnost in mutacijo genoma«, ki sta bili skupaj temeljno vključeni v aktiviranje osmih značilnih (funkcionalnih) zmožnosti, potrebnih za rast in napredovanje tumorja. Res je, da ta pregled vključuje dodatne predlagane nove značilnosti in omogočitvene funkcije, vključno z »odklepanjem fenotipske plastičnosti«, »nemutacijskim epigenetskim reprogramiranjem«, »polimorfnimi mikrobiomi« in »starečimi celicami«. Blagovni znamki grafike raka sta bili prevzeti od Hanahana in Weinberga (2).
Kot smo takrat omenili, same te značilne značilnosti ne morejo obravnavati kompleksnosti patogeneze raka, tj. natančne molekularne in celične mehanizme, ki omogočajo razvijajočim se preneoplastičnim celicam, da razvijejo in pridobijo te nenormalne fenotipske sposobnosti med potekom tumorigeneze in malignega napredovanja.
Skladno s tem smo v razpravo dodali še en koncept, ki je predstavljen kot »mogoče lastnosti«, posledice nenormalnega stanja neoplazme, ki zagotavlja sredstva, s katerimi lahko rakave celice in tumorji pridobijo te funkcionalne značilnosti. Kot take se omogočitvene lastnosti odražajo v molekularnih in celičnih mehanizmih, preko katerih se pridobijo značilnosti, ne pa v samih zgornjih osmih veščinah. Ta dva procesa aktivacije sta bila nestabilnost genoma in vnetje, ki spodbuja tumor.
Nadalje smo ugotovili, da tumorsko mikrookolje (TME), ki je tu opredeljeno kot sestavljeno iz heterogenih in interaktivnih populacij rakavih celic in matičnih celic raka skupaj z različnimi vrstami rekrutiranih stromalnih celic – transformirani parenhim in z njim povezana stroma – je zdaj splošno cenjeno, da igra bistveno vlogo pri tumorigenezi in malignem napredovanju.
Glede na stalno zanimanje za te formulacije in naš nadaljnji namen, da spodbujamo stalno razpravo in izboljšanje sheme Hallmarks, je primerno razmisliti o pogosto zastavljenem vprašanju: Ali obstajajo dodatne značilnosti tega konceptualnega modela, ki bi jih bilo mogoče vključiti, ob upoštevanju potrebe po zagotovitvi tega? da so široko uporabni v celotnem spektru človeških rakov? V skladu s tem predstavljam več potencialnih novih značilnosti in omogočitvenih funkcij, ki bi jih lahko sčasoma vključili kot ključne komponente značilnosti konceptualizacije raka.
Ti parametri so "odklepanje fenotipske plastičnosti", "nemutacijsko epigenetsko reprogramiranje", "polimorfni mikrobiomi" in "stareče celice" (slika 1, desno). Pomembno je, da so primeri, predstavljeni v podporo tem tezam, ilustrativni, a nikakor ne izčrpni, saj obstaja vedno več in vse bolj prepričljivih objavljenih dokazov, ki podpirajo vsako vinjeto.
Določanje fenotipske plastičnosti
Med organogenezo razvoj, določitev in organizacijo celic v tkiva za opravljanje homeostatskih funkcij spremlja končna diferenciacija, pri čemer matične celice prenehajo rasti, včasih nepovratno, ko ti procesi dosežejo vrhunec. Kot tak je končni rezultat celične diferenciacije v večini primerov antiproliferativen, kar tvori jasno oviro za nadaljnjo proliferacijo, potrebno za neoplazijo.
Vse več je dokazov, da je sprostitev običajno omejene sposobnosti fenotipske plastičnosti za izogibanje stanju terminalne diferenciacije kritična sestavina patogeneze raka (3). Ta plastičnost lahko deluje v več manifestacijah (slika 2). Tako lahko nastajajoče rakave celice, ki izvirajo iz normalne celice, ki se je razvila po poti, ki se približuje ali prevzame popolnoma diferencirano stanje, obrnejo smer z dediferenciacijo nazaj v stanja celic, podobnih matičnim.
Nasprotno pa lahko neoplastične celice, ki izhajajo iz matične celice, ki sledi poti, ki vodi do končne diferenciacije, povzročijo kratek stik in vzdržujejo rastoče rakave celice v delno diferenciranem stanju, podobnem matičnim celicam. Alternativno lahko pride do transdiferenciacije, pri kateri celice, ki so bile prvotno zavezane eni diferenciacijski poti, preklopijo na popolnoma drugačen razvojni program in s tem pridobijo tkivno specifične značilnosti, ki niso bile vnaprej določene z njihovimi normalnimi izvornimi celicami.
Naslednji primeri podpirajo trditev, da različne oblike celične plastičnosti razkrivajo fenotipsko plastičnost. Na levi je fenotipska plastičnost nedvomno pridobljena značilna sposobnost, ki omogoča različne motnje diferenciacije celic, vključno z (i) dediferenciacijo od zrelih do matičnih stanj, (ii) zastalo (terminalno) diferenciacijo iz stanj matičnih celic in (iii) transdiferenciacijo v druge celične linije. Na desni so prikazani trije vidni načini oslabljene diferenciacije, ki so sestavni del patogeneze raka.
Z različnim sprevračanjem normalne diferenciacije matičnih celic v zrele celice v razvojnih linijah se olajšata tumorigeneza in maligno napredovanje, ki izhaja iz izvornih celic v takih poteh. Blagovni znamki grafike raka sta bili prevzeti od Hanahana in Weinberga (2).
Slika 2

Dediferenciacija
Karcinogeneza debelega črevesa je primer oslabljene diferenciacije, saj obstaja teleološka potreba po začetnih rakavih celicah, da se izognejo tekočemu traku končne diferenciacije in luščenja, kar bi se načeloma lahko zgodilo z dediferenciacijo epitelijskih celic debelega črevesa, ki se še niso končno diferencirale, ali prek zastale diferenciacije progenitorskih/matičnih celic v kriptah, ki povzročajo te diferenciacijske celice. Tako diferencirane celice kot izvorne celice so bile vpletene v celice izvora raka debelega črevesa (4–6).
Dva razvojna transkripcijska faktorja (TF), homeobox protein HOXA5 in SMAD4, slednji vključen v signalizacijo BMP, sta močno izražena pri diferenciaciji epitelijskih celic debelega črevesa in se običajno izgubita pri napredovalih karcinomih debelega črevesa, ki značilno izražajo markerje matičnih in matičnih celic. Funkcionalne motnje v mišjih modelih so pokazale, da prisilna ekspresija HOXA5 v rakavih celicah debelega črevesa obnovi označevalce diferenciacije, zavira fenotipe matičnih celic in zmanjša invazijo in metastaze, kar daje utemeljitev za njegovo značilno znižanje regulacije (7, 8).
Nasprotno pa SMAD4 uveljavlja diferenciacijo in zatiranje proliferacije, ki jo poganja onkogena signalizacija WNT, kar se razkrije z inženirsko izgubo izražanja SMAD4, kar zagotavlja razlago za njegovo izgubo izražanja, da se omogoči dediferenciacija in posledično hiperproliferacija, ki jo poganja WNT (5).
Predvsem je izguba teh dveh "zaviralcev diferenciacije" s posledično dediferenciacijo povezana s pridobitvijo drugih sposobnosti zaznamka, kot tudi drugih regulatorjev, ki inducirajo zaznamek, kar otežuje strogo opredelitev tega začasnega zaznamka kot ločljivega in neodvisnega.
Drugi dokazi se nanašajo na potlačeno izražanje glavnega regulatorja diferenciacije melanocitov MITF, za katerega se zdi, da je vpleten v nastanek agresivnih oblik malignega melanoma. Izguba tega razvojnega TF je povezana z reaktivacijo progenitorskih genov nevralnega grebena in znižano regulacijo genov, ki so značilni za popolnoma diferencirane melanocite. Ponovna pojavnost genov nevralnega grebena kaže, da se te celice vrnejo v prvotno stanje, iz katerega razvojno izhajajo melanociti.
Poleg tega je študija sledenja liniji melanomov, ki jih povzroča BRAF, ugotovila zrele pigmentirane melanocite kot izvorne celice, ki so podvržene dediferenciaciji med potekom tumorigeneze (9). Predvsem mutirani onkogen BRAF, ki ga najdemo v več kot polovici kožnih melanomov, inducira hiperproliferacijo, ki je pred in jo je zato mogoče mehanično ločiti od kasnejše dediferenciacije, ki izhaja iz znižane regulacije MITF.
Druga študija je funkcionalno implicirala uravnavanje razvojnega TF ATF2, katerega značilno izražanje v mišjih in človeških melanomih posredno zavira MITF1, sočasno z malignim napredovanjem posledično dediferenciranih celic melanoma (10). Nasprotno pa izražanje v melanomih mutantnih oblik ATF2, ki ne morejo zatreti MITF, povzroči dobro diferencirane melanome (11).
Poleg tega je nedavna študija (12) povezala dediferenciacijo linije z malignim napredovanjem neoplazem pankreasnih otočkov v karcinome, ki so nagnjeni k metastazam; te nevroendokrine celice in iz njih izpeljani tumorji izvirajo iz razvojne linije, ki se razlikuje od tiste, ki ustvarja veliko večje število sosednjih celic, ki tvorijo eksokrine celice in trebušno slinavko ter nastale duktalne adenokarcinome.
Zanimivo je, da je bila pot večstopenjske diferenciacije od matičnih celic otočkov do zrelih β-celic temeljito opisana (13). Primerjalno profiliranje transkriptoma kaže, da so adenomu podobni tumorji otočkov najbolj podobni nezrelim, a diferenciranim β-celicam, ki proizvajajo insulin, medtem ko so invazivni karcinomi najbolj podobni prekurzorjem embrionalnih otočkov. Napredovanje v slabo diferencirane karcinome vključuje začetni korak dediferenciacije, ki na začetku ne vključuje povečane proliferacije ali zmanjšane apoptoze v primerjavi z dobro diferenciranimi adenomi, ki se oba ponavadi pojavita pozneje.
Tako diskretnega koraka dediferenciacije ne poganjajo opazne spremembe v značilnih značilnostih trajne proliferacije in odpornosti na apoptozo. Namesto tega je regulacija miRNA, ki je bila prej vpletena v določanje matičnega stanja otočkov, tista, ki je znižana med terminalno diferenciacijo β-celic, 12).
Blokirana diferenciacija
Medtem ko zgornji primeri ponazarjajo, kako lahko supresija ekspresije diferenciacijskega faktorja olajša tumorigenezo, tako da omogoči bolje diferenciranim celicam, da se dediferencirajo v matične celice, lahko v drugih primerih nepopolno diferencirane matične celice utrpijo regulativne spremembe, ki aktivno blokirajo njihovo nadaljnje napredovanje v popolnoma diferencirana, tipično neproliferativna stanja.
Že dolgo je dokumentirano, da je akutna promielocitna levkemija (APL) posledica kromosomske translokacije, ki spoji lokus PML z genom, ki kodira jedrski receptor retinojske kisline α (RARα). Mieloidne matične celice, ki nosijo take translokacije, očitno ne morejo nadaljevati svoje običajne terminalne diferenciacije v granulocite, kar ima za posledico celice, ujete v proliferativno, promielocitom podobno fazo matičnih celic (14).
Dokaz koncepta za to shemo izhaja iz zdravljenja gojenih celic APL, mišjih modelov bolezni in prizadetih bolnikov z retinojsko kislino, ligandom RARα; To terapevtsko zdravljenje povzroči, da se neoplastične APL celice diferencirajo v navidezno zrele, neproliferirajoče granulocite, s čimer prekinejo njihovo progresivno proliferativno širjenje (14–16).
Različica na to temo se nanaša na drugo obliko akutne mieloične levkemije, ki nosi translokacijo t(8;21), ki proizvaja fuzijski protein AML1-ETO. Samo ta protein lahko transformira mieloidne progenitorje, vsaj deloma z blokiranjem njihove diferenciacije. Terapevtska intervencija pri mišjih modelih in bolnikih s farmakološkim zaviralcem histonske deacetilaze, ki spreminja kromatin (HDAC), povzroči, da se celice mieloične levkemije ponovno diferenciirajo v celice z bolj zrelo morfologijo mieloidnih celic. To reakcijo spremlja zmanjšanje proliferativne sposobnosti, kar ovira napredovanje te levkemije (17, 18).
Tretji primer pri melanomu vključuje razvojni TF, SOX10, ki je med diferenciacijo melanocitov običajno znižan. Študije pridobitve in izgube funkcije v modelu melanomov, ki jih povzroča BRAF, so pokazale, da nenormalno vzdrževana ekspresija SOX10 blokira diferenciacijo nevralnih matičnih celic v melanocite, kar omogoča nastanek melanomov, ki jih povzroča BRAF (19).
Drugi primeri modulatorjev diferenciacije vključujejo metabolit alfa-ketoglutarat (αKG), potreben kofaktor za številne encime, ki spreminjajo kromatin, za katerega se je izkazalo, da sodeluje pri spodbujanju določenih diferenciranih stanj celic. Pri raku trebušne slinavke tumorski supresor p53 stimulira nastajanje αKG in vzdrževanje bolj diferenciranega celičnega stanja, medtem ko prototipna izguba funkcije p53 vodi do znižanja ravni αKG in posledične dediferenciacije, ki je povezana z malignim napredovanjem (20).
Pri eni od oblik jetrnega raka mutacija gena za izocitrat dehidrogenazo (IDH1/2) ne povzroči nastajanja αKG, ki povzroča diferenciacijo, temveč sorodnega »onkometabolita«, D-2-hidroksigluterata (D2HG), za katerega se je pokazalo, da blokira diferenciacijo hepatocitov matičnih celic jeter prek represije glavnega regulatorja, ki ga posreduje D2HG. diferenciacija in mirovanje hepatocitov, HNF4a.
Z D2HG posredovana supresija funkcije HNF4a sproži proliferativno ekspanzijo progenitornih celic hepatocitov v jetrih, ki postanejo dovzetne za onkogeno transformacijo ob kasnejši mutacijski aktivaciji onkogena KRAS, kar požene maligno napredovanje v holangiokarcinom jeter (21). Mutant IDH1/2 in njegov onkometabolit D2HG delujeta tudi pri različnih vrstah mieloidnih in drugih solidnih tumorjev, kjer D2HG zavira αKG-odvisne dioksigenaze, ki so potrebne za dogodke metilacije histona in DNA, ki posredujejo spremembe v strukturi kromatina med diferenciacijo razvojne linije, s čimer zamrznejo nastanek rakavih celic v progenitornem stanju (22, 23).
Dodaten, soroden koncept je »obhodna diferenciacija«, pri kateri delno ali nediferencirane matične/progenitorne celice zapustijo celični cikel in mirujejo v zaščitnih nišah, s potencialom za ponovno sprožitev proliferativne ekspanzije (24), čeprav še vedno s selektivnim pritiskom, da se na tak ali drugačen način prekine njihova programirana diferenciacija.
Transdiferenciacija
Koncept transdiferenciacije so patologi že dolgo prepoznali v obliki tkivne metaplazije, pri kateri celice določenega diferenciranega fenotipa izrazito spremenijo svojo morfologijo, da postanejo jasno prepoznavne kot elementi drugega tkiva, vidni primer tega je Barrettov požiralnik, kjer kronično vnetje večplastnega skvamoznega epitelija požiralnika inducira transdiferenciacijo v preprost stolpčasti epitelij, značilen za črevesje, s čimer olajša kasnejši razvoj adenokarcinomov namesto ploščatoceličnih karcinomov, pričakovanih od tega ploščatoceličnega epitelija (3).
Zdaj molekularne determinante razkrivajo mehanizme transdiferenciacije pri različnih oblikah raka, tako za primere, kjer je groba tkivna metaplazija očitna, kot za druge, kjer je nekoliko bolj subtilna, kot ponazarjajo naslednji primeri.
Informativni primer transdiferenciacije kot diskretnega dogodka v tumorigenezi se nanaša na duktalni adenokarcinom trebušne slinavke (PDAC), pri katerem se lahko ena od vpletenih izvornih celic, acinarna celica trebušne slinavke, transdiferencira v fenotip duktalne celice med začetkom razvoja neoplastike. Dva TF-ja – PTF1a in MIST1 – nadzirata specifikacijo in vzdrževanje diferenciranega stanja acinarnih celic trebušne slinavke prek njihovega izražanja v kontekstu samozadostnih regulacijskih zank »naprej« (25).
Oba TF sta pogosto znižana med neoplastičnim razvojem in malignim napredovanjem človeškega in mišjega PDAC. Funkcionalne genetske študije na miših in kultiviranih človeških celicah PDAC so pokazale, da eksperimentalno vsiljena ekspresija PTF1a poslabša transdiferenciacijo in proliferacijo, ki jo povzroči KRAS, in lahko tudi prisili ponovno diferenciacijo že neoplastičnih celic v mirujoči fenotip acinarnih celic (26).
Nasprotno pa supresija ekspresije PTF1a sproži metaplazijo acinarja v kanal, in sicer transdiferenciacijo, in s tem senzibilizira kanalu podobne celice za onkogeno transformacijo KRAS, kar pospeši poznejši razvoj invazivnega PDAC (27). Podobno prisilna ekspresija MIST1 v trebušni slinavki, ki izraža KRAS, prav tako blokira transdiferenciacijo in poslabša začetek tumorigeneze trebušne slinavke, ki je sicer olajšana s tvorbo premalignih kanalom podobnih lezij (PanIN), medtem ko genetska delecija MIST1 poveča njihovo tvorbo in začetek neoplastičnega napredovanja, ki ga poganja KRAS (28).
Izguba izražanja PTF1 ali MIST1 med tumorigenezo je povezana s povečanim izražanjem drugega razvojnega regulatornega TF, SOX9, ki je običajno učinkovit pri specifikaciji duktalnih celic (27, 28). Prisilna regulacija SOX9, s čimer se izognemo potrebi po znižani regulaciji PTF1a in MIST1, se je prav tako pokazalo, da spodbuja transdiferenciacijo acinarnih celic v fenotip duktalnih celic, občutljiv na neoplazijo, ki jo povzroča KRAS (29), kar pomeni, da je SOX9 ključni funkcionalni efektor njihove znižane regulacije v genezi človeškega PDAC.
Tako lahko tri TF, ki uravnavajo diferenciacijo trebušne slinavke, spremenimo na različne načine, da induciramo transdiferencirano stanje, ki v kontekstu mutacijske aktivacije KRAS olajša onkogeno transformacijo ter začetek tumorigeneze in malignega napredovanja.
Dodatni člani družine SOX regulacijskih faktorjev, povezanih s kromatinom, so po eni strani v veliki meri povezani tako s specifikacijo celične usode kot s preklapljanjem rodov v razvoju (30), po drugi strani pa z več fenotipi, povezanimi s tumorji (31). Drug pomemben primer transdiferenciacije, posredovane s SOX, vključuje mehanizem terapevtske odpornosti pri raku prostate.
V tem primeru je izguba tumorskih supresorjev RB in p53 - katerih odsotnost je značilna za nevroendokrine tumorje - kot odgovor na antiandrogeno terapijo potrebna, vendar ne zadostna za običajno opaženo transformacijo dobro diferenciranih celic raka prostate v celice karcinoma, ki so vdrle v linijo diferenciacije z molekularnimi in histološkimi značilnostmi nevroendokrinih celic, ki zlasti ne izražajo androgenega receptorja. Poleg izgube RB in p53 pridobljena odpornost na antiandrogensko terapijo zahteva povečano izražanje SOX2, razvojnega regulatornega gena, za katerega se je izkazalo, da pomaga inducirati transdiferenciacijo celic adenokarcinoma, ki se odzivajo na terapijo, v derivate, ki so v stanju nevroendokrinih celic, neodzivnih na terapijo (32).
Tretji primer prav tako prikazuje transdiferenciacijo kot strategijo, ki jo uporabljajo celice karcinoma, da bi se izognile odstranitvi s terapijo, specifično za rod, v tem primeru z bazalnoceličnimi karcinomi (BCC) kože, zdravljene s farmakološkim zaviralcem onkogene poti Hedgehog-Smoothened (HH/SMO), za katero je znano, da spodbuja neoplastično rast teh celic (33).
Rakave celice, odporne na zdravila, preidejo v razvojno povezano, vendar ločeno celično vrsto s širokimi epigenetskimi premiki v specifičnih domenah kromatina in spremenjeno dostopnostjo dveh superojačevalcev. Novo pridobljeno fenotipsko stanje celic BCC jim omogoča, da ohranijo izražanje onkogene signalne poti WNT, kar posledično daje neodvisnost od signalne poti HH/SMO, zatrte z zdravili (34).
Kot je bilo pričakovano od te transdiferenciacije, se transkriptom rakavih celic premakne iz genskega podpisa, ki odraža vpleteno celico izvora BCC, in sicer izvornih celic izbokline lasnega mešička, v podpis, ki kaže na bazalne matične celice, ki naseljujejo interfolikularno povrhnjico BCC. Takšna transdiferenciacija, ki omogoča odpornost na zdravila, je vse bolj dokumentirana pri različnih oblikah raka (35).
Zdi se, da je plastičnost razvojne linije prevladujoča tudi pri glavnih podtipih pljučnega karcinoma, tj. pri nevroendokrinih karcinomih [drobnocelični pljučni rak (SCLC)] in adenokarcinomi + ploščatocelični karcinomi [zbirni nedrobnocelični pljučni rak (NSCLC)]. Sekvenciranje enocelične RNA je razkrilo izjemno dinamično in heterogeno pretvorbo med temi podtipi, kot tudi izrazite razlike med stopnjami pljučne tumorigeneze, kasnejšega malignega napredovanja in odziva na terapijo (36-38).
Zato namesto preproste konceptualizacije čistega klonskega prehoda iz ene linije v drugo te študije slikajo veliko bolj zapleteno sliko dinamično medsebojno pretvarjajočih se subpopulacij rakavih celic, ki kažejo značilnosti več razvojnih linij in diferenciacijskih stopenj, kar je trezen vpogled v zvezi s tem za terapevtsko ciljanje raka pljuč pri ljudeh na podlagi rodov. Začenjajo se ugotavljati regulativne determinante te dinamične fenotipske plastičnosti (37, 39, 40).
Povzetek
Trije zgoraj opisani razredi mehanizmov poudarjajo selektivne regulatorje celične plastičnosti, ki jih je – vsaj delno – mogoče ločiti od osrednjih onkogenih gonilnikov in drugih značilnih zmožnosti. Poleg teh primerov obstaja veliko dokazov, ki povezujejo številne oblike raka z oslabljeno diferenciacijo, ki jo spremlja pridobivanje podpisov transkriptoma in drugih fenotipov - na primer histološke morfologije - ki so povezani s stopnjami prednikov ali izvornih celic, opaženimi v ustreznih normalnih tkivih. izvora ali v drugih bolj oddaljenih vrstah celic in linijah (41–43).
Kot taki se zdi, da so ti trije podrazredi fenotipske plastičnosti – dediferenciacija zrelih celic nazaj v matična stanja, zastala diferenciacija za zamrznitev razvijajočih se celic v stanjih matičnih celic in transdiferenciacija v alternativne celične linije – učinkoviti pri več vrstah raka med primarno tumorigenezo, malignim napredovanjem in/ali odzivom na terapijo.
Vendar obstajata dva konceptualna vidika. Prvič, dediferenciacija in zastala diferenciacija sta verjetno prepleteni, saj ju ni mogoče razlikovati pri mnogih vrstah tumorjev, v katerih izvorna celica - diferencirana celica ali matična celica - ni znana ali je alternativno vključena. Drugič, pridobivanje ali ohranjanje fenotipov matičnih celic in izguba diferenciranih lastnosti je v večini primerov netočen odraz normalne razvojne stopnje, ki potopi v okolje drugih značilnih sprememb v rakavi celici, ki niso prisotne v naravno razvijajočih se celicah.
Poleg tega druga oblika fenotipske plastičnosti vključuje celično staranje, ki je na splošno obravnavano spodaj, pri čemer lahko rakave celice, ki so podvržene navidezno nepovratnemu staranju, namesto tega pobegnejo in nadaljujejo s proliferativno ekspanzijo (44). Nazadnje, tako kot pri drugih značilnih zmožnostih, celična plastičnost ni nov izum ali aberacija rakavih celic, temveč korupcija latentnih, a aktivirajočih se zmožnosti, ki jih različne normalne celice uporabljajo za podporo homeostaze, popravljanja in regeneracije (45).
Na splošno ti ilustrativni primeri spodbujajo k razmisleku, da sprostitev celične plastičnosti za omogočanje različnih oblik motene diferenciacije predstavlja izrazito razlikovalno sposobnost, ki se v regulaciji in celičnem fenotipu razlikuje od dobro potrjenih osnovnih značilnosti raka (slika 2).
Epigenetsko reprogramiranje brez mutacije
Omogočena lastnost nestabilnosti in mutacije genoma (DNK) je temeljna sestavina razvoja in patogeneze raka. Trenutno več mednarodnih konzorcijev katalogizira mutacije v celotnem genomu človeških rakavih celic, v skoraj vseh vrstah človeškega raka, na različnih stopnjah malignega napredovanja, vključno z metastatskimi lezijami, in med razvojem odpornosti na prilagodljivo terapijo. Eden od rezultatov je zdaj splošno razširjeno spoznanje, da se mutacije v genih, ki organizirajo, modulirajo in vzdržujejo kromatinsko arhitekturo in s tem uravnavajo izražanje genov na svetovni ravni, vedno bolj odkrivajo in funkcionalno povezujejo z lastnostmi raka (46–48).
Poleg tega obstajajo argumenti za drugo na videz neodvisno obliko reprogramiranja genoma, ki vključuje čisto epigenetsko regulirane spremembe v izražanju genov, takšno, ki bi jo lahko poimenovali »nemutacijsko epigenetsko reprogramiranje« (slika 3). Pravzaprav je bila teza o evoluciji raka brez mutacij in izključno epigenetskem programiranju značilnih fenotipov raka postavljena pred skoraj desetletjem (49) in se o njej čedalje bolj razpravlja (46, 50–52).
Slika 3

Podobno kot se dogaja med embriogenezo ter diferenciacijo in homeostazo tkiv, kopičenje dokazov kaže, da lahko instrumentalna genska regulacijska vezja in mreže v tumorjih nadzorujejo množica pokvarjenih in kooptiranih mehanizmov, ki so neodvisni od nestabilnosti genoma in genske mutacije. Blagovni znamki grafike raka sta bili prevzeti od Hanahana in Weinberga (2).
Seveda je koncept nemutacijske epigenetske regulacije izražanja genov dobro uveljavljen kot osrednji mehanizem, ki posreduje embrionalni razvoj, diferenciacijo in organogenezo (53-55). Pri odraslih, na primer, dolgoročni spomin vključuje spremembe v modifikaciji genov in histonov, v strukturi kromatina in v proženju stikal za izražanje genov, ki se skozi čas stabilno ohranjajo s pozitivnimi in negativnimi povratnimi zankami (56, 57). Vse več dokazov podpira idejo, da lahko analogne epigenetske spremembe prispevajo k pridobitvi značilnih sposobnosti med razvojem tumorja in malignim napredovanjem. V podporo tej hipotezi je spodaj predstavljenih nekaj primerov.
Mikrookoljski mehanizmi epigenetskega reprogramiranja
Kako se reprogramira genom rakave celice, če ne samo z onkogenimi mutacijami? Vse več dokazov kaže, da lahko aberantne fizikalne lastnosti tumorskega mikrookolja povzročijo široke spremembe v epigenomu, od katerih lahko spremembe, ki so koristne za fenotipsko izbiro lastnosti lastnosti, vodijo do klonskega razraščanja rakavih celic z izboljšano sposobnostjo za proliferativno širjenje.
Skupna značilnost tumorjev (ali regij znotraj tumorjev) je hipoksija kot posledica neustrezne vaskularizacije. Hipoksija, na primer, zmanjša aktivnost demetilaz TET, kar povzroči pomembne spremembe v metilomu, zlasti hipermetilacijo (58). Nezadostna vaskularizacija bo verjetno tudi omejila biološko uporabnost kritičnih hranil, ki se prenašajo s krvjo, in pomanjkanje hranil, na primer, je pokazalo, da spremeni translacijski nadzor in posledično poveča maligni fenotip celic raka dojke (59).
Prepričljiv primer epigenetske regulacije, ki jo posreduje hipoksija, je oblika vedno smrtonosnega pediatričnega ependimoma. Tako kot mnogi embrionalni in pediatrični tumorji tudi tej obliki manjkajo ponavljajoče se mutacije, zlasti pomanjkanje pogonskih mutacij v onkogenih in zaviralcih tumorja. Namesto tega se je izkazalo, da je nenormalna rast teh rakavih celic nadzorovana s hipoksijo induciranim genskim regulatornim programom (60, 61). Zanimivo je, da domnevna celica izvora tega raka prebiva v hipoksičnem predelu in verjetno senzibilizira celice v njem, da sprožijo tumorigenezo prek še neznanih kofaktorjev.
Še en prepričljiv dokaz za epigenetsko regulacijo, posredovano z mikrookoljem, se nanaša na sposobnost invazivne rasti rakavih celic. Klasičen primer je reverzibilna indukcija invazivnosti rakavih celic na robovih številnih solidnih tumorjev, ki jo orkestrira razvojni regulativni program, znan kot prehod epitelija v mezenhim (EMT; ref. 62–64). Predvsem je bilo nedavno dokazano, da glavni regulator EMT, ZEB1, inducira ekspresijo histonske metiltransferaze, SETD1B, ki nato ohranja ekspresijo ZEB1 v pozitivni povratni zanki, ki vzdržuje (invazivno) regulativno stanje EMT (65).
Prejšnja študija je podobno dokumentirala, da je indukcija EMT s povečano ekspresijo povezanega TF, SNAIL1, povzročila izrazite spremembe v pokrajini kromatina kot rezultat indukcije številnih modifikatorjev kromatina, katerih aktivnost se je izkazala za potrebno za vzdrževanje fenotipskega stanja (66). Poleg tega lahko številni pogoji in dejavniki, ki jih doživljajo rakave celice na robovih tumorjev, vključno s hipoksijo in citokini, ki jih izločajo stromalne celice, očitno inducirajo EMT in s tem invazivnost (67, 68).
Osupljiv primer programiranja invazivnosti z mikrookoljem, ki domnevno ni povezan s programom EMT, vključuje avtokrino aktivacijo nevronskega signalnega vezja, ki vključuje izločeni glutamat in njegov receptor NMDAR (69, 70). Zanimivo je, da ima prototipna togost mnogih solidnih tumorjev, utelešena v obsežnih spremembah zunajceličnega matriksa (ECM), ki obdaja celice v njih, globoke posledice za invazivne in druge fenotipske lastnosti rakavih celic.
V primerjavi z običajnim tkivnim ECM, iz katerega izhajajo tumorji, je tumorski ECM značilno povečano navzkrižno povezovanje in gostota, encimske modifikacije in spremenjena molekularna sestava, ki skupaj orkestrirajo, delno preko integrinskih receptorjev za motive ECM, s togostjo povzročeno signalizacijo in genske ekspresijske mreže, ki inducirajo invazivnost in druge značilne značilnosti (71).
Poleg takšnih regulativnih mehanizmov, ki jih zagotavlja fizično mikrookolje tumorja, lahko parakrino signaliziranje, ki vključuje topne dejavnike, ki jih v zunajcelično okolje sproščajo različni tipi celic, ki naseljujejo solidne tumorje, prispeva tudi k indukciji več morfološko različnih invazivnih programov rasti (72), od katerih se zdi, da je samo eden – imenovan »mezenhimski« – vključen v zgoraj omenjeno epigenetsko regulativni mehanizem EMT.
Epigenetska regulativna heterogenost
Rastoča baza znanja povečuje spoštovanje pomena intratumoralne heterogenosti pri ustvarjanju fenotipske raznolikosti, kjer najprimernejše celice za proliferativno širjenje in invazijo prerastejo svoje brate in so zato izbrane za maligno napredovanje. Vsekakor je en vidik te fenotipske heterogenosti posledica kronične ali epizodne genomske nestabilnosti in posledične genetske heterogenosti v celicah, ki naseljujejo tumor.
Poleg tega postaja vse bolj jasno, da lahko obstaja epigenetska heterogenost, ki ne temelji na mutacijah. Izrazit primer je povezovalni histon H1.0, ki se dinamično izraža in potlači v subpopulacijah rakavih celic znotraj vrste tumorjev, s posledično sekvestracijo ali dostopnostjo megabaznih domen [73]. Predvsem je bilo ugotovljeno, da ima populacija rakavih celic z potlačenim H1.0 lastnosti, podobne steblu, izboljšano sposobnost iniciacije tumorja in povezavo s slabo prognozo pri bolnikih.
Drug primer epigenetsko regulirane plastičnosti je bil opisan pri človeških peroralnih skvamoznoceličnih karcinomih (SCC), kjer rakave celice na invazivnih robovih prevzamejo delno stanje EMT (p-EMT), ki nima zgoraj omenjenih mezenhimskih TF, vendar izraža druge gene, ki določajo EMT, ki niso izraženi v osrednjem jedru tumorjev (74).
Celice p-EMT očitno ne predstavljajo klonske kompartmentalizacije mutacijsko spremenjenih celic: kulture rakavih celic, pridobljenih iz primarnega tumorja, vsebujejo dinamične mešanice celic p-EMT hi in p-EMT lo in ko so bile celice p-EMT hi/lo prečiščene in kultivirane s FACS, sta se obe vrnili v mešane populacije p-EMT hi in p-EMT lo v 4 dneh. Čeprav bi parakrine signale iz sosednje strome lahko šteli za deterministične za p-EMT hi stanje, stabilna prisotnost in regeneracija obeh epigenetskih stanj v kulturi govorita o intrinzičnem mehanizmu rakavih celic. Predvsem je ta zaključek podprt z analizo 198 celičnih linij, ki predstavljajo 22 vrst raka, vključno s SCC, kjer je bilo 12 stabilno heterogenih epigenetskih stanj (vključno s p-EMT v SCC) različno odkritih v modelih celičnih linij kot tudi z njimi povezanimi primarnimi tumorji (75).
Spet heterogenih fenotipskih stanj ni bilo mogoče povezati z zaznavnimi genetskimi razlikami in v več primerih se je pokazalo, da se celice določenega stanja, razvrščene po FACS, dinamično ponovno uravnovesijo po kulturi, kar povzema stabilno ravnovesje med heterogenimi stanji, opaženimi v prvotnih celičnih linijah.
Poleg tega tehnologije za celotno genomsko profiliranje različnih atributov – poleg zaporedja DNK in njegove mutacijske variacije – osvetljujejo vplivne elemente opombe in organizacije genoma rakave celice, ki so v korelaciji s prognozo bolnika in vse bolj z značilnimi sposobnostmi (76–78). Epigenomsko heterogenost razkrivajo vse zmogljivejše tehnologije za profiliranje metilacije DNA v celotnem genomu (79, 80), modifikacije histonov (81), dostopnosti kromatina (82) ter post-transkripcijske modifikacije in prevoda RNA (83, 84).
Izziv v zvezi s tukaj obravnavanim postulatom bo določiti, katere epigenomske spremembe v določenih vrstah raka (i) imajo regulativni pomen in (ii) so reprezentativne za povsem nemutacijsko reprogramiranje, v nasprotju z mutacijo, ki jo poganja in s tem z genomom razložljiva nestabilnost.
Epigenetska regulacija vrst stromalnih celic, ki naseljujejo tumorsko mikrookolje
Na splošno se domneva, da pomožne celice v tumorskem mikrookolju, ki funkcionalno prispevajo k pridobivanju značilnih sposobnosti, ne trpijo zaradi genetske nestabilnosti in mutacijskega reprogramiranja, da bi okrepile svoje dejavnosti, ki spodbujajo tumor; namesto tega se sklepa, da so te celice – fibroblasti, povezani z rakom, prirojene imunske celice in endotelijske celice ter periciti tumorske vaskulature – epigenetično reprogramirane po njihovi rekrutaciji s topnimi in fizikalnimi dejavniki, ki definirajo mikrookolje trdnega tumorja (2, 85).
Pričakuje se, da se bodo multi-omske tehnologije profiliranja, ki se trenutno uporabljajo za rakave celice, vedno bolj uporabljale za preučevanje pomožnih (stromalnih) celic v tumorjih, da bi razjasnili, kako so normalne celice poškodovane, da funkcionalno podpirajo razvoj in napredovanje tumorja. Na primer, nedavna študija (86) kaže, da lahko takšno reprogramiranje vključuje modifikacije epigenoma, poleg induktivne izmenjave citokinov, kemokinov in rastnih faktorjev, ki spreminjajo znotrajcelična signalna omrežja v vseh teh vrstah celic:
Ko so mišje modele z metastazami na pljučih zdravili s kombinacijo zaviralca DNA metiltransferaze (5-azacitidina) in zaviralca modifikacije histona (HDAC), je bilo ugotovljeno, da so infiltrirane mieloidne celice prešle iz nezrelega matičnega stanja (ki spodbuja tumor) v celice, ki spominjajo na zrele intersticijske (nasprotujejo tumorju) makrofage, ki za razliko od svojih dvojnikov v nezdravljeni tumorji, niso mogli podpreti tipičnih zmogljivosti, potrebnih za učinkovito metastatsko kolonizacijo (86). Možno je, da bodo multi-omično profiliranje in farmakološke motnje služile za pojasnitev reprogramiranega epigenetskega stanja v takšnih mieloidnih celicah kot tudi drugih značilnih tipih pomožnih celic, ki poseljujejo tumorska mikrookolja.
Povzetek
Ti ilustrativni posnetki skupaj podpirajo tezo, da bo epigenetsko reprogramiranje brez mutacije sprejeto kot prava lastnost, ki omogoča, da olajša pridobivanje značilnih sposobnosti (slika 3), ki se razlikujejo od nestabilnosti in mutacije genomske DNK. Zlasti lahko pričakujemo, da se bo nemutacijsko epigenetsko reprogramiranje izkazalo za sestavni del omogočanja predhodne nove razlikovalne sposobnosti fenotipske plastičnosti, o kateri smo razpravljali zgoraj, zlasti kot gonilna sila v dinamični transkriptomski heterogenosti, ki je vse bolj dobro dokumentirana v TME malignih rakavih celic. Napredek enoceličnih multi-omičnih tehnologij profiliranja bo osvetlil zadevne prispevke in medsebojno delovanje med epigenetsko regulacijo, ki jo poganja mutacija, in ne, ki jo poganja mutacija, pri razvoju tumorjev med malignim napredovanjem in metastazami.
Polimorfni mikrobiomi
Daljnosežna meja v biomedicini se odpira z osvetljevanjem raznolikosti in variabilnosti številčnosti mikroorganizmov, ki jih skupaj imenujemo mikrobiota, ki se simbiotično povezujejo s pregradnimi tkivi telesa, izpostavljenimi zunanjemu okolju - zlasti povrhnjici in notranji sluznici prebavil, pa tudi pljuč, dojk in genitourinarnega sistema.
Vse bolj se priznava, da imajo ekosistemi, ki jih ustvarijo rezidenčne bakterije in glive – mikrobiomi – globoke učinke na zdravje in bolezni ( 87 ), spoznanje, ki ga poganja sposobnost pregledovanja populacij mikrobnih vrst z uporabo sekvenciranja naslednje generacije in bioinformatičnih tehnologij. Za raka postajajo vedno bolj prepričljivi dokazi, da ima lahko polimorfna variabilnost mikrobiomov med posamezniki v populaciji globoke učinke na fenotipe raka (88, 89).
Asociacijske študije pri ljudeh in eksperimentalne manipulacije na mišjih modelih raka razkrivajo določene mikroorganizme, predvsem, a ne izključno bakterije, ki imajo lahko zaščitne ali škodljive učinke na razvoj raka, maligno napredovanje in odziv na terapijo. To velja tudi za globalno kompleksnost in sestavo tkivnega mikrobioma kot celote. Medtem ko je bil črevesni mikrobiom začetnik te nove meje, ima več tkiv in organov povezane mikrobiome, ki kažejo posebne lastnosti, povezane s populacijsko dinamiko in raznolikostjo mikrobnih vrst in podvrst.
To vse večje spoštovanje pomena polimorfno variabilnih mikrobiomov v zdravju in bolezni postavlja vprašanje: Ali je mikrobiom izrazita lastnost, ki omogoča, da na splošno pozitivno in negativno vpliva na pridobitev značilnih sposobnosti za raka? To možnost obravnavam spodaj in ponazarjam dokaze za nekatere vidne tkivne mikrobiome, ki so vpleteni v lastnosti raka (slika 4), začenši z najvidnejšim in očitno najbolj vplivnim mikrobiomom, mikrobiomom črevesnega trakta.
Slika 4
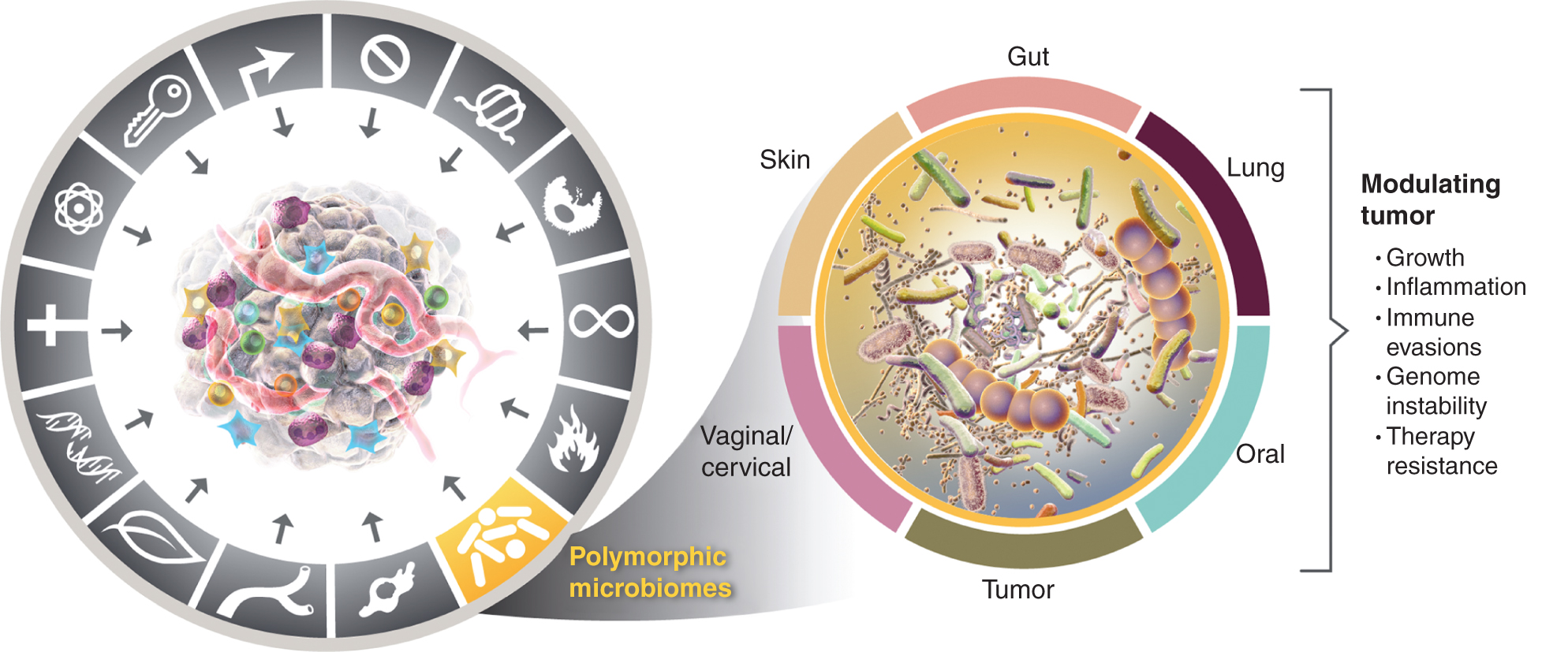
Levo, medtem ko se omogočitvene lastnosti vnetja, ki spodbuja tumor, ter genomske nestabilnosti in mutacije prekrivajo, je vedno več razlogov za sklepanje, da lahko polimorfni mikrobiomi, ki se nahajajo pri enem posamezniku v primerjavi z drugim v debelem črevesu, v drugih sluznicah in povezanih organih ali v samih tumorjih, vplivajo na številne značilne sposobnosti na različne načine – bodisi z indukcijo ali inhibicijo – in so zato lahko instrumentalni in skoraj neodvisni spremenljivka v uganki o tem, kako se rak razvija, napreduje in raste, odziva na terapijo. Res je, da je več tkivnih mikrobiomov vključenih v modulacijo tumorskih fenotipov. Poleg široko raziskanega črevesnega mikrobioma so drugi značilni tkivni mikrobiomi in tumorski mikrobiom vključeni v modulacijo pridobitve - tako pozitivne kot negativne - značilnih sposobnosti, predstavljenih v določenih vrstah tumorjev. Blagovni znamki grafike raka sta bili prevzeti od Hanahana in Weinberga (2).
Več modulacijskih učinkov črevesnega mikrobioma
Že dolgo je znano, da je črevesni mikrobiom temeljnega pomena za delovanje debelega črevesa (debelega črevesa) pri razgradnji in uvažanju hranilnih snovi v telo kot del presnovne homeostaze in da lahko motnje mikrobnih populacij – disbioza – v debelem črevesu povzročijo spekter fizioloških bolezni (87). To vključuje sum, da na dovzetnost, razvoj in patogenezo raka debelega črevesa vpliva črevesni mikrobiom. V zadnjih letih so prepričljive funkcionalne študije z uporabo fekalnih presadkov bolnikov s tumorjem debelega črevesa in miši v miši prejemnice, nagnjene k razvoju raka debelega črevesa, vzpostavile načelo: obstajajo mikrobiomi, ki ščitijo pred rakom in spodbujajo tumorje, ki vključujejo specifične bakterijske vrste, ki lahko modulirajo pojav in patogenezo tumorjev debelega črevesa (90).
Mehanizmi, s katerimi mikrobiota podeljuje te modulatorne vloge, so še vedno pojasnjeni, vendar sta dva splošna učinka vse bolj uveljavljena za mikrobiome, ki spodbujajo tumorje, in v nekaterih primerih za specifične bakterijske vrste, ki spodbujajo tumor. Prvi učinek je mutageneza epitelija debelega črevesa kot posledica proizvodnje bakterijskih toksinov in drugih molekul, ki neposredno poškodujejo DNK ali motijo sisteme, ki ohranjajo genomsko celovitost ali drugače obremenjujejo celice, kar posredno vpliva na zvestobo podvajanja in popravljanja DNK. Tipičen primer je E. coli, ki nosi lokus PKS, ki dokazano mutagenira človeški genom in sodeluje pri prenosu mutacij, ki omogočajo znamenje (91).
Poleg tega so poročali, da se bakterije vežejo na površino epitelijskih celic debelega črevesa in proizvajajo mimetike ligandov, ki stimulirajo proliferacijo epitelija, kar prispeva k značilni proliferativni signalni sposobnosti v neoplastičnih celicah (88). Drug mehanizem, s katerim posebne vrste bakterij spodbujajo razvoj tumorja, so bakterije, ki proizvajajo butirat, katerih številčnost je povečana pri bolnikih s kolorektalnim rakom (92).
Proizvodnja metabolita butirata ima zapletene fiziološke učinke, vključno z indukcijo starajočih se epitelijskih in fibroblastnih celic. Mišji model karcinogeneze debelega črevesa, koloniziran z bakterijami, ki proizvajajo butirat, je razvil več tumorjev kot miši brez takih bakterij; Povezava med staranjem, povzročenim z butiratom, in povečano tumorigenezo debelega črevesa je bila dokazana z uporabo senolitičnega zdravila, ki uniči starajoče se celice in zmanjša rast tumorja (92).
Poleg tega ima bakterijsko proizveden butirat pleiotropne in paradoksalne učinke na diferencirane celice v primerjavi z nediferenciranimi (matičnih) celicami v epiteliju debelega črevesa v pogojih, kjer je črevesna pregrada porušena (disbioza) in so bakterije invazivne ter vplivajo na primer na celično energijo in metabolizem, modifikacijo histonov, napredovanje celičnega cikla in (ki spodbuja tumor) prirojeni imunski vnetje, ki imunosupresira adaptivne imunske odzive (93).
Široko delovanje polimorfnih mikrobiomov dejansko vključuje modulacijo adaptivnih in prirojenih imunskih sistemov po različnih poteh, vključno s proizvodnjo "imunomodulatornih" faktorjev s strani bakterij, ki aktivirajo senzorje poškodb na epitelijskih ali rezidenčnih imunskih celicah, kar vodi do izražanja raznolikega repertoarja kemokinov in citokinov, ki lahko oblikujejo številčnost in lastnosti imunskih celic, ki naseljujejo epitelija debelega črevesa in spodnje ležeče strome ter drenažnih bezgavk.
Poleg tega lahko nekatere bakterije zlomijo tako zaščitni biofilm kot sluz, ki obdaja epitelij debelega črevesa, in prekinejo tesne stike med epitelnimi celicami in celicami, ki skupaj ohranjajo celovitost fizične pregrade, ki običajno deli črevesni mikrobiom. Po vdoru v stromo lahko bakterije sprožijo tako prirojene kot adaptivne imunske odzive, tako da izzovejo izločanje repertoarja citokinov in kemokinov. Ena od manifestacij je lahko ustvarjanje imunskega mikrookolja, ki spodbuja ali tumorju nasprotuje, kar posledično ščiti ali olajša nastanek tumorjev in maligno napredovanje.
V skladu s tem je lahko modulacija prepletenih parametrov (i) indukcije (prirojenega) vnetja, ki spodbuja tumor, in (ii) pobega pred (adaptivnim) imunskim uničenjem s strani značilnih mikrobiomov pri posameznih bolnikih povezana ne le z prognozo, temveč tudi z odzivom ali odpornostjo na imunoterapije z zaviralci imunskih kontrolnih točk in drugimi terapevtskimi načini (ena od manifestacij je lahko ustvarjanje imunsko mikrookolje, ki pospešuje ali tumorju nasprotuje, ki posledično nastanejo Zaščitijo ali olajšajo razvoj tumorja in maligno napredovanje.
V skladu s tem je lahko modulacija prepletenih parametrov (i) indukcije (prirojenega) vnetja, ki spodbuja tumor, in (ii) pobega pred (adaptivnim) imunskim uničenjem s strani značilnih mikrobiomov pri posameznih bolnikih povezana ne le z prognozo, temveč tudi z odzivom ali odpornostjo na imunoterapije z zaviralci imunskih kontrolnih točk in drugimi terapevtskimi načini (ena od manifestacij je lahko ustvarjanje imunsko mikrookolje, ki spodbuja ali tumorju nasprotuje, ki posledično nastanejo, zaščitijo ali olajšajo razvoj tumorja in maligno napredovanje).
V skladu s tem je lahko modulacija prepletenih parametrov (i) indukcije (prirojenega) vnetja, ki spodbuja tumor, in (ii) pobega pred (adaptivnim) imunskim uničenjem z značilnimi mikrobiomi pri posameznih bolnikih povezana ne le z prognozo, ampak tudi z odzivom ali odpornostjo na imunoterapije z zaviralci imunskih kontrolnih točk in drugimi terapevtskimi modalitetami (89, 94–96). Preliminarni dokaz koncepta izhaja iz nedavnih študij, ki kažejo na obnovljeno učinkovitost imunoterapije po presaditvi fekalne mikrobiote iz bolnikov, ki so se odzvali na terapijo, v bolnike z melanomom, ki je napredoval med predhodnim zdravljenjem z blokado imunske kontrolne točke (97, 98).
Molekularni mehanizmi, s katerimi različne in spremenljive komponente črevesnega mikrobioma sistemsko modulirajo aktivnost adaptivnega imunskega sistema, ostajajo vztrajna skrivnost, bodisi s krepitvijo protitumorskih imunskih odzivov, ki jih izzove blokada imunske kontrolne točke, bodisi z indukcijo sistemske ali lokalne (intratumoralne) imunosupresije. Nedavna študija je osvetlila: nekateri sevi Enterococcus (in drugih bakterij) izražajo peptidoglikansko hidroliazo, imenovano SagA, ki sprošča mukopeptide iz bakterijske stene, ki lahko nato sistemsko krožijo in aktivirajo receptor vzorca NOD2, kar posledično poveča odziv T-celic in učinkovitost imunoterapije na kontrolnih točkah (99).
Druge imunoregulacijske molekule, ki jih proizvajajo specifične bakterijske podvrste, so identificirane in funkcionalno ocenjene, vključno z bakterijsko proizvedenim inozinom, presnovkom, ki omejuje hitrost za aktivnost celic T (100). Ti in drugi primeri začenjajo razmejevati molekularne mehanizme, s katerimi polimorfni mikrobiomi posredno in sistemsko modulirajo tumorsko imunobiologijo, poleg imunskih odzivov, ki sledijo neposrednim fizičnim interakcijam bakterij z imunskim sistemom (101, 102).
Poleg vzročnih povezav z rakom debelega črevesa in melanomom je dokazana sposobnost mikrobioma črevesja, da izzove izražanje imunomodulatornih kemokinov in citokinov, ki vstopajo v sistemski krvni obtok, očitno tudi sposobna vplivati na patogenezo raka in odziv na terapije v drugih telesnih organih (94, 95).
Razsvetljujoč primer zadeva razvoj holangiokarcinomov v jetrih: črevesna disbioza omogoča vstop in transport bakterij in bakterijskih produktov skozi portalno veno v jetra, kjer se sproži TLR4, izražen na hepatocitih, da inducira ekspresijo kemokina CXCL1, ki rekrutira granulocitne mieloidne celice (gMDSC), ki izražajo CXCR2 in služijo zatiranju naravne celice ubijalke, da se izognejo imunskemu uničenju (103) in verjetno prenašajo druge značilne sposobnosti (85). Kot tak je črevesni mikrobiom jasno impliciran kot omogočitvena lastnost, ki lahko alternativno olajša ali zaščiti pred več vrstami raka.
Onkraj črevesja: impliciranje različnih mikrobiomov v drugih pregradnih tkivih
Skoraj vsa tkiva in organi, ki so neposredno ali posredno izpostavljeni zunanjemu okolju, so tudi skladišča komenzalnih mikroorganizmov (104). V nasprotju s črevesjem, kjer je simbiotska vloga mikrobioma v presnovi dobro prepoznana, se normalne in patogene vloge rezidenčne mikrobiote na teh različnih lokacijah še vedno pojavljajo.
Obstajajo očitne razlike, značilne za organ/tkivo, v konstituciji povezanih mikrobiomov v homeostazi, staranju in raku, pri čemer se tako prekrivajo kot značilne vrste in pogostnosti glede na tiste v debelem črevesu (104, 105). Poleg tega asociacijske študije zagotavljajo vedno več dokazov, da lahko lokalni tkivni mikrobiomi, ki nasprotujejo/zaščitijo tumor, v primerjavi s tkivnimi mikrobiomi, ki spodbujajo tumor, podobno kot črevesni mikrobiom, modulirajo dovzetnost in patogenezo za raka pri ljudeh, ki nastanejo v njihovih povezanih organih (106–109).
Vpliv intratumorske mikrobiote?
Nazadnje, patologi že dolgo priznavajo, da je mogoče bakterije odkriti v solidnih tumorjih, opažanje, ki je bilo zdaj utemeljeno s sofisticiranimi tehnologijami profiliranja. Na primer, v študiji 1.526 tumorjev, ki zajemajo sedem vrst človeškega raka (kosti, možgani, dojke, pljuča, melanom, jajčniki in trebušna slinavka), je bila za vsako vrsto značilen poseben mikrobiom, ki se večinoma nahaja v rakavih celicah in imunskih celicah. Znotraj vsakega tipa tumorja so bile dokazane razlike v tumorskem mikrobiomu in zaključeno, da so povezane s klinično-patološkimi značilnostmi (110).
Mikrobiota je bila podobno odkrita v de novo genetsko spremenjenih mišjih modelih pljučnega raka in raka trebušne slinavke, njihova odsotnost pri miših brez mikrobov in/ali njihova razveljavitev z antibiotiki pa lahko poslabša tumorigenezo, kar funkcionalno implicira tumorski mikrobiom kot predhodnika vnetja, ki spodbuja tumor, in malignega napredovanja (111, 112).
Asociacijske študije pri duktalnem adenokarcinomu trebušne slinavke pri ljudeh in funkcionalni testi s transplantacijo iztrebkov v miši s tumorjem so pokazale, da variacije tumorskega mikrobioma – in z njim povezanega mikrobioma črevesja – modulirajo fenotipe in preživetje imunskega sistema (113). Pomemben izziv za prihodnost bo razširiti te posledice na druge vrste tumorjev in ločiti potencialno ločljive prispevke konstitucije in variacije mikrobioma tumorja od mikrobioma črevesja (in lokalnega tkiva izvora), morda z identifikacijo specifičnih mikrobnih vrst, ki so funkcionalno vplivne na enem ali drugem mestu.
Povzetek
Zanimiva vprašanja za prihodnost vključujejo, ali lahko mikrobiota, ki prebiva v različnih tkivih ali naseljuje začetne neoplazme, prispeva k ali moti pridobitev drugih značilnih zmožnosti, ki presegajo imunomodulacijo in genomsko mutacijo, s čimer vpliva na razvoj in napredovanje tumorja. Obstajajo dokazi, da lahko nekatere bakterijske vrste neposredno stimulirajo značilnost proliferativne signalizacije, na primer v epiteliju debelega črevesa (88), in lahko modulirajo zaviranje rasti s spreminjanjem aktivnosti supresorja tumorja v različnih predelih črevesja (114), medtem ko neposredni učinki na druge značilne sposobnosti, kot je izogibanje celični smrti, sprožitev angiogeneze ter spodbujanje invazije in metastaz, ostajajo. nejasna, kot tudi posplošljivost teh opažanj na več oblik človeškega raka.
Ne glede na to obstajajo vedno bolj prepričljivi argumenti, da polimorfne variacije v mikrobiomih črevesja in drugih organov predstavljajo značilno aktivacijsko lastnost za pridobivanje značilnih veščin (slika 4), čeprav se prekrivajo in dopolnjujejo tiste nestabilnosti in mutacije genoma ter vnetja, ki spodbujajo tumor.
Stare celice
Celično staranje je tipično ireverzibilna oblika ustavitve proliferacije, ki se je verjetno razvila kot zaščitni mehanizem za vzdrževanje homeostaze tkiva, navidezno kot dopolnilni mehanizem programirani celični smrti, ki služi za inaktivacijo in pravočasno odstranitev obolelih, disfunkcionalnih ali drugače nepotrebnih celic. Poleg zaustavitve cikla celične delitve program staranja povzroči spremembe v celični morfologiji in metabolizmu ter, najbolj globoko, aktivacijo s staranjem povezanega sekretornega fenotipa (SASP), ki vključuje sproščanje množice bioaktivnih beljakovin, vključno s kemokini.
Citokini in proteaze, katerih identiteta je odvisna od vrste celice in tkiva, iz katerega nastane starajoča se celica (115–117). Staranje lahko v celicah povzročijo različni pogoji, vključno s stresi v mikrookolju, kot sta pomanjkanje hranil in poškodbe DNK, pa tudi poškodbe organelov in celične infrastrukture ter neravnovesja v celičnih signalnih omrežjih (115, 117), ki so se vsi pojavili v kontekstu opaženega povečanja pogostosti starajočih se celic v različnih organih med staranjem (118, 119).
Celično staranje je dolgo veljalo za zaščitni mehanizem pred neoplazijo, ki povzroča staranje rakavih celic (120). Večina zgoraj omenjenih sprožilcev programa staranja je povezana z malignostjo, predvsem s poškodbo DNA kot posledico aberantne hiperproliferacije, tako imenovano onkogeno inducirano senescenco zaradi hiperaktivirane signalizacije in terapevtsko inducirano senescenco kot posledico celične in genomske poškodbe zaradi kemoterapije in radioterapije.
Dejansko obstajajo dobro uveljavljeni primeri zaščitnih koristi staranja pri omejevanju malignega napredovanja (118, 119). Nasprotno pa vedno več dokazov kaže ravno nasprotno: v določenih okoliščinah starajoče se celice različno spodbujajo razvoj tumorja in maligno napredovanje (119, 121).
V pronicljivi študiji primera so bile starajoče se celice pri starajočih miših farmakološko ablirane, pri čemer so bile posebej izčrpane starajoče se celice, ki značilno izražajo inhibitor celičnega cikla p16 – INK4a: poleg odložitve več s starostjo povezanih simptomov je to povzročilo izčrpavanje starajočih se celic pri starajočih se miših z zmanjšano pojavnostjo spontane tumorigeneze in smrti, povezane z rakom (122).
Glavni mehanizem, s katerim starajoče se celice spodbujajo tumorske fenotipe, naj bi bil SASP, za katerega se je izkazalo, da lahko posreduje signalne molekule (in proteaze, ki se aktivirajo in/ali deaktivirajo) na parakrini način za posredovanje tipičnih zmožnosti. Tako se je v različnih eksperimentalnih sistemih pokazalo, da stare rakave celice na različne načine prispevajo k proliferativni signalizaciji, izogibajo se apoptozi, inducirajo angiogenezo, spodbujajo invazijo in metastaze ter zavirajo imunost tumorja (116, 118, 120, 121).
Še en vidik učinkov starajočih se rakavih celic na fenotipe raka vključuje prehodna, reverzibilna stanja starajočih se celic, pri čemer se lahko starajoče rakave celice izognejo svojemu neproliferativnemu stanju, v katerem se izraža SASP, ter nadaljujejo s celično proliferacijo in manifestacijo povezanih zmožnosti popolnoma sposobnih onkogenih celic (44).
Tako prehodno staranje je najbolje dokumentirano v primerih odpornosti na terapijo (44), ki predstavlja obliko mirovanja, ki se izogne terapevtskemu ciljanju na proliferirajoče rakave celice, vendar se lahko izkaže za širšo učinkovitost v drugih fazah razvoja tumorja, malignega napredovanja in metastaz.
Poleg tega sposobnosti starajočih se celic, ki spodbujajo znak, niso omejene na starajoče se rakave celice. Pokazalo se je, da fibroblasti, povezani z rakom (CAF), starajo v tumorjih, kar povzroča staranje CAF, za katere se je izkazalo, da spodbujajo tumor, tako da podeljujejo značilne sposobnosti rakavim celicam v TME (115, 116, 121).
Poleg tega so starajoči se fibroblasti v normalnih tkivih, delno oblikovani z naravnim staranjem ali okoljskimi obremenitvami, podobno vključeni v preoblikovanje tkivnega mikrookolja prek svojega SASP, da zagotovijo parakrino podporo za lokalno invazijo (tako imenovani »učinki polja«) in oddaljene metastaze (116) neoplazem, ki se razvijajo v bližini.
Poleg tega se je pokazalo, da starajoči se fibroblasti v starajoči se koži rekrutirajo – prek svojega SASP – prirojene imunske celice, ki imunosupresivno delujejo na adaptivne protitumorske imunske odzive, zasidrane s celicami CD8 T, in spodbujajo rast kožnega tumorja (123), pri čemer slednji učinek verjetno odraža parakrine prispevke takšnih prirojenih imunskih celic (mieloidnih celic, nevtrofilcev in makrofagi) odraža druge značilne sposobnosti.
Čeprav je manj dobro uveljavljeno, se zdi verjetno, da bodo druge obilne stromalne celice, ki poseljujejo specifično tumorsko mikrookolje, podvržene staranju, s čimer bodo modulirale značilnosti raka in posledične tumorske fenotipe. Na primer, endotelijske celice starega tumorja, ki jih povzroči terapija, lahko povečajo proliferacijo, invazijo in metastaze v modelih raka dojke (124, 125).
Vsekakor takšni dokazi upravičujejo preiskavo pri drugih tipih tumorjev, da bi ocenili splošno staranje fibroblastov, endotelijskih celic in drugih stromalnih celic kot gonilne sile pri razvoju tumorja. Prav tako so trenutno nejasni regulativni mehanizmi in funkcionalne determinante, s katerimi določen tip starajoče se celice v določenem TME izzove SASP, ki spodbuja tumor, v primerjavi s tumorsko antagonizirajočim SASP, ki se očitno lahko alternativno inducira v istem tipu starajoče se celice, morda z različnimi iniciatorji, ko so potopljeni v značilna fiziološka in neoplastična mikrookolja.
Povzetek
Koncept, da so tumorji sestavljeni iz gensko spremenjenih rakavih celic, ki medsebojno delujejo in imajo koristi od rekrutiranih in epigenetsko/fenotipsko pokvarjenih pomožnih (stromalnih) celic, je bil uveljavljen kot ključen za patogenezo raka. Premisleki, obravnavani zgoraj in opisani v pregledih in poročilih, citiranih tukaj (in drugje), prepričljivo dokazujejo, da je treba starejše celice (ne glede na celični izvor) upoštevati pri vključitvi na seznam funkcionalno pomembnih celic v tumorskem mikrookolju (slika 5). Zato je treba pri iskanju poglobljenega znanja o mehanizmih raka upoštevati starajoče se celice. Poleg tega priznanje njihovega pomena motivira sekundarni cilj terapevtskega ciljanja na starajoče se celice, ki spodbujajo tumor, vseh konstitucij, bodisi s farmakološko ali imunološko ablacijo ali z reprogramiranjem SASP v različice, ki nasprotujejo tumorju (115, 121, 126).
Slika 5

Heterogeni podtipi rakavih celic ter tipi in podtipi stromalnih celic so funkcionalno integrirani v manifestacije tumorjev kot nezakonitih organov. Vse več dokazov kaže, da so derivati starajočih se celic mnogih od teh celičnih komponent TME in njihovih spremenljivih SASP vključeni v modulacijo značilnih zmogljivosti in posledičnih tumorskih fenotipov. Blagovni znamki grafike raka sta bili prevzeti od Hanahana in Weinberga (2).
Sklepne opombe
Medtem ko se je izkazalo, da ima osem značilnih znakov raka in njihovi dve podporni značilnosti trajno hevristično vrednost pri konceptualizaciji raka, zgoraj predstavljeni premisleki kažejo, da morda obstajajo novi vidiki nekaj splošnega in zato pomembni za popolnejše razumevanje zapletenosti, mehanizmov in manifestacij bolezni. Z uporabo metrike opazne, če ne popolne neodvisnosti od 10 osnovnih lastnosti, je mogoče trditi, da se lahko ti štirje parametri – po nadaljnji validaciji in posplošitvi, ki presega predstavljene študije primerov – vključijo v značilnosti sheme raka (slika 6).
Zato bi celično plastičnost lahko dodali na seznam izstopajočih zmogljivosti. Medtem ko se osmo jedro in ta nova sposobnost konceptualno razlikujeta po svoji definiciji kot značilnosti, so vidiki njune regulacije vsaj delno povezani pri nekaterih in morda številnih vrstah raka. Na primer, več znakov je koordinirano moduliranih s kanoničnimi onkogenimi gonilniki v nekaterih vrstah tumorjev, vključno z
- (I) KRAS ( https://cancer.sanger.ac.uk/cosmic/census-page/KRAS ),
- (II) MYC ( https://cancer.sanger.ac.uk/cosmic/census-page/MYC ),
- (III) NOTCH ( https://cancer.sanger.ac.uk/cosmic/census-page/NOTCH1 ; Ref. 127) und
- (IV) TP53 ( https://cancer.sanger.ac.uk/cosmic/census-page/TP53 )
Slika 6
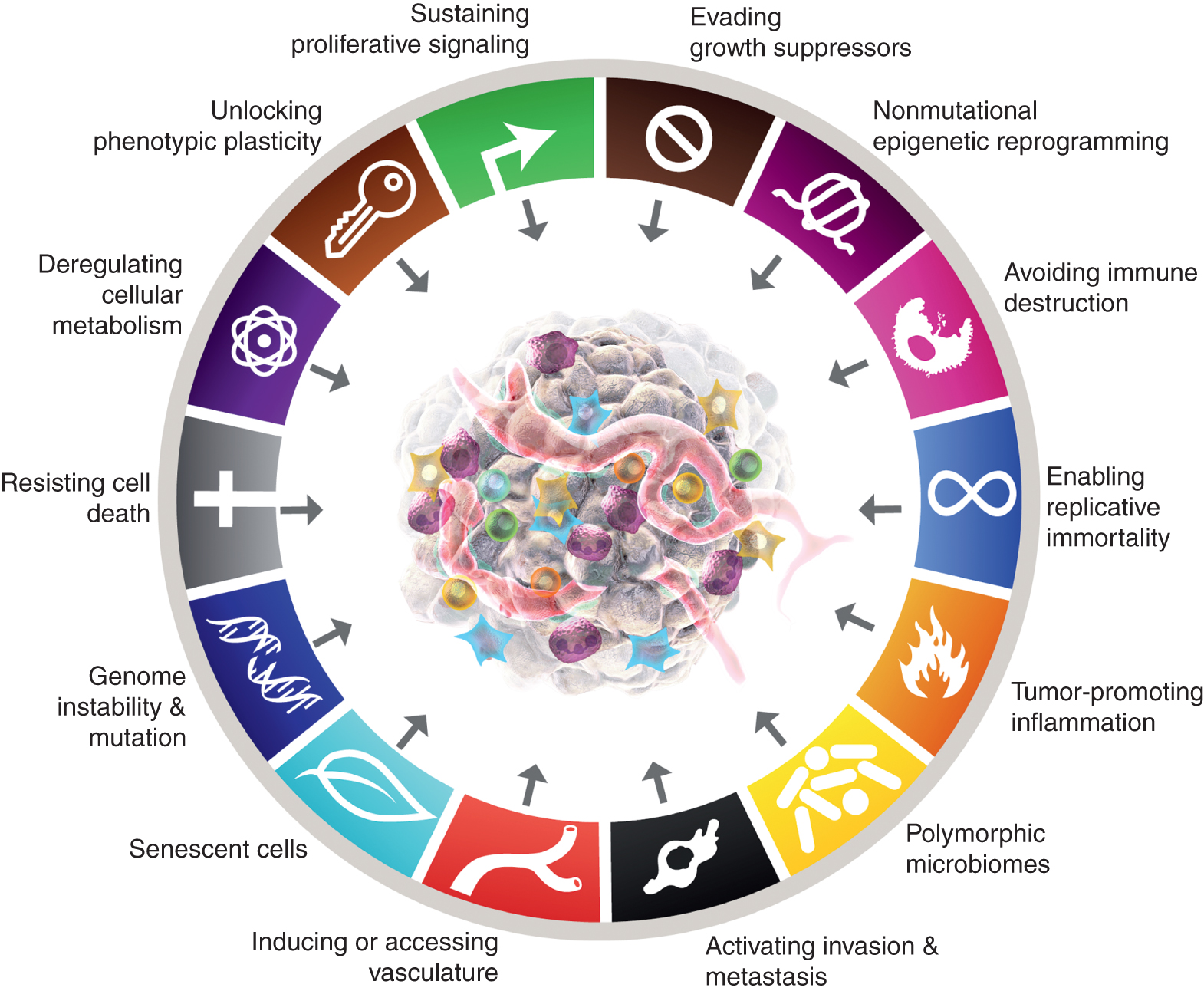
Prikazani so kanonični in pričakovani novi dodatki k »Znakom raka«. Ta članek postavlja možnost, da bi spodbudili razpravo, razpravo in eksperimentalno izpopolnjevanje, da bodo nekateri ali vsi od štirih novih parametrov priznani kot generični za več oblik človeškega raka in zato primerni za integracijo v temeljno konceptualizacijo znakov raka. Blagovni znamki grafike raka sta bili prevzeti od Hanahana in Weinberga (2).
Poleg dodajanja celične plastičnosti na seznam se lahko nemutacijsko epigenetsko reprogramiranje in polimorfne variacije integrirajo v mikrobiome organov/tkiv kot mehanične determinante – omogočitvene lastnosti – prek katerih se pridobijo značilne zmožnosti, skupaj z vnetjem, ki spodbuja tumor (ki je samo delno povezano z mikrobiomom), poleg mutacij in drugih aberacij, ki kažejo omenjene onkogene dejavnike. zgoraj.
Nazadnje, senescentne celice različnega izvora - vključno z rakavimi celicami in različnimi stromalnimi celicami - ki funkcionalno prispevajo k razvoju in malignemu napredovanju raka, čeprav na izrazito drugačen način od tistih svojih nestarečih bratov, se lahko vključijo kot generične komponente TME. Če povzamemo, predvideva se, da bo uporaba teh preliminarnih "eksperimentalnih balonov" spodbudila razpravo, razpravo in nadaljnje eksperimentalne raziskave v skupnosti raziskovalcev raka o definiranju konceptualnih parametrov biologije, genetike in patogeneze raka.
Reference
- Hanahan D , Weinberg RA . The hallmarks of cancer. Cell 2000;100:57–70.
- Hanahan D , Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Yuan S , Norgard RJ , Stanger BZ . Cellular plasticity in cancer. Cancer Discov 2019;9:837–51.
- Barker N , Ridgway RA , van Es JH , van de Wetering M , Begthel H , van den Born M et al . Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608–11.
- Perekatt AO , Shah PP , Cheung S , Jariwala N , Wu A , Gandhi V et al . SMAD4 suppresses WNT-driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res 2018;78:4878–90.
- Shih IM , Wang TL , Traverso G , Romans K , Hamilton SR , Ben-Sasson S et al . Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A 2001;98:2640–5.
- Ordóñez-Morán P , Dafflon C , Imajo M , Nishida E , Huelsken J . HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 2015;28:815–29.
- Tan SH , Barker N . Stemming colorectal cancer growth and metastasis: HOXA5 forces cancer stem cells to differentiate. Cancer Cell 2015;28:683–5.
- Köhler C , Nittner D , Rambow F , Radaelli E , Stanchi F , Vandamme N et al . Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 2017;21:679–93.
- Shah M , Bhoumik A , Goel V , Dewing A , Breitwieser W , Kluger H et al . A role for ATF2 in regulating MITF and melanoma development. PLoS Genet 2010;6:e1001258.
- Claps G , Cheli Y , Zhang T , Scortegagna M , Lau E , Kim H et al . A transcriptionally inactive ATF2 variant drives melanomagenesis. Cell Rep 2016;15:1884–92.
- Saghafinia S , Homicsko K , Di Domenico A , Wullschleger S , Perren A , Marinoni I et al . Cancer cells retrace a stepwise differentiation program during malignant progression. Cancer Discov 2021;11:2638–57.
- Yu X-X , Qiu W-L , Yang L , Zhang Y , He M-Y , Li L-C et al . Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J 2019;38:e100164.
- de Thé H . Differentiation therapy revisited. Nat Rev Cancer 2018;18:117–27.
- He LZ , Merghoub T , Pandolfi PP . In vivo analysis of the molecular pathogenesis of acute promyelocytic leukemia in the mouse and its therapeutic implications. Oncogene 1999;18:5278–92.
- Warrell RP , de Thé H , Wang ZY , Degos L . Acute promyelocytic leukemia. N Engl J Med 1993;329:177–89.
- Bots M , Verbrugge I , Martin BP , Salmon JM , Ghisi M , Baker A et al . Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014;123:1341–52.
- Ferrara FF , Fazi F , Bianchini A , Padula F , Gelmetti V , Minucci S et al . Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res 2001;61:2–7.
- Kaufman CK , Mosimann C , Fan ZP , Yang S , Thomas AJ , Ablain J et al . A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016;351:aad2197.
- Morris JP , Yashinskie JJ , Koche R , Chandwani R , Tian S , Chen C-C et al . α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019;573:595–9.
- Saha SK , Parachoniak CA , Ghanta KS , Fitamant J , Ross KN , Najem MS et al . Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110–4.
- Dang L , Su S-SM . Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem 2017;86:305–31.
- Waitkus MS , Diplas BH , Yan H . Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018;34:186–95.
- Phan TG , Croucher PI . The dormant cancer cell life cycle. Nat Rev Cancer 2020;20:398–411.
- Jiang M , Azevedo-Pouly AC , Deering TG , Hoang CQ , DiRenzo D , Hess DA et al . MIST1 and PTF1 collaborate in feed-forward regulatory loops that maintain the pancreatic acinar phenotype in adult mice. Mol Cell Biol 2016;36:2945–55.
- Krah NM , Narayanan SM , Yugawa DE , Straley JA , Wright CVE , MacDonald RJ et al . Prevention and reversion of pancreatic tumorigenesis through a differentiation-based mechanism. Dev Cell 2019;50:744–54.
- Krah NM , De La O J-P , Swift GH , Hoang CQ , Willet SG , Chen Pan F et al . The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015;4:e07125.
- Shi G , DiRenzo D , Qu C , Barney D , Miley D , Konieczny SF . Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 2013;32:1950–8.
- Kopp JL , von Figura G , Mayes E , Liu F-F , Dubois CL , Morris JP et al . Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50.
- Julian LM , McDonald AC , Stanford WL . Direct reprogramming with SOX factors: masters of cell fate. Curr Opin Genet Dev 2017;46:24–36.
- Grimm D , Bauer J , Wise P , Krüger M , Simonsen U , Wehland M et al . The role of SOX family members in solid tumours and metastasis. Semin Cancer Biol 2020;67:122–53.
- Mu P , Zhang Z , Benelli M , Karthaus WR , Hoover E , Chen C-C et al . SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84–8.
- Von Hoff DD , LoRusso PM , Rudin CM , Reddy JC , Yauch RL , Tibes R et al . Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72.
- Biehs B , Dijkgraaf GJP , Piskol R , Alicke B , Boumahdi S , Peale F et al . A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018;562:429–33.
- Boumahdi S , de Sauvage FJ . The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19:39–56.
- Groves SM , Ireland A , Liu Q , Simmons AJ , Lau K , Iams WT et al . Cancer Hallmarks Define a Continuum of Plastic Cell States between Small Cell Lung Cancer Archetypes [Internet]. Systems Biology; 2021 Jan. Available from: http://biorxiv.org/lookup/doi/10.1101/2021.01.22.427865.
- LaFave LM , Kartha VK , Ma S , Meli K , Del Priore I , Lareau C et al . Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 2020;38:212–28.
- Marjanovic ND , Hofree M , Chan JE , Canner D , Wu K , Trakala M et al . Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 2020;38:229–46.
- Drapkin BJ , Minna JD . Studying lineage plasticity one cell at a time. Cancer Cell 2020;38:150–2.
- Inoue Y , Nikolic A , Farnsworth D , Liu A , Ladanyi M , Somwar R et al . Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer [Internet]. Cancer Biology; 2020 Nov. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.11.12.368522.
- Dravis C , Chung C-Y , Lytle NK , Herrera-Valdez J , Luna G , Trejo CL et al . Epigenetic and transcriptomic profiling of mammary gland development and tumor models disclose regulators of cell state plasticity. Cancer Cell 2018;34:466–82.
- Malta TM , Sokolov A , Gentles AJ , Burzykowski T , Poisson L , Weinstein JN et al . Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–54.
- Miao Z-F , Lewis MA , Cho CJ , Adkins-Threats M , Park D , Brown JW et al . A dedicated evolutionarily conserved molecular network licenses differentiated cells to return to the cell cycle. Dev Cell 2020;55:178–94.
- De Blander H , Morel A-P , Senaratne AP , Ouzounova M , Puisieux A . Cellular plasticity: a route to senescence exit and tumorigenesis. Cancers 2021;13:4561.
- Merrell AJ , Stanger BZ . Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 2016;17:413–25.
- Baylin SB , Jones PA . Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 2016;8:a019505.
- Flavahan WA , Gaskell E , Bernstein BE . Epigenetic plasticity and the hallmarks of cancer. Science 2017;357:eaal2380.
- Jones PA , Issa J-PJ , Baylin S . Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630–41.
- Huang S . Tumor progression: Chance and necessity in Darwinian and Lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol 2012;110:69–86.
- Darwiche N . Epigenetic mechanisms and the hallmarks of cancer: an intimate affair. Am J Cancer Res 2020;10:1954–78.
- Feng Y , Liu X , Pauklin S . 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell 2021;12:440–54.
- Nam AS , Chaligne R , Landau DA . Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet 2021;22:3–18.
- Bitman-Lotan E , Orian A . Nuclear organization and regulation of the differentiated state. Cell Mol Life Sci CMLS 2021;78:3141–58.
- Goldberg AD , Allis CD , Bernstein E . Epigenetics: a landscape takes shape. Cell 2007;128:635–8.
- Zeng Y , Chen T . DNA methylation reprogramming during mammalian development. Genes 2019;10:257.
- Hegde AN , Smith SG . Recent developments in transcriptional and translational regulation underlying long-term synaptic plasticity and memory. Learn Mem 2019;26:307–17.
- Kim S , Kaang B-K . Epigenetic regulation and chromatin remodeling in learning and memory. Exp Mol Med 2017;49:e281.
- Thienpont B , Van Dyck L , Lambrechts D . Tumors smother their epigenome. Mol Cell Oncol 2016;3:e1240549.
- Gameiro PA , Struhl K . Nutrient deprivation elicits a transcriptional and translational inflammatory response coupled to decreased protein synthesis. Cell Rep 2018;24:1415–24.
- Lin GL , Monje M . Understanding the deadly silence of posterior fossa A ependymoma. Mol Cell 2020;78:999–1001.
- Michealraj KA , Kumar SA , Kim LJY , Cavalli FMG , Przelicki D , Wojcik JB et al . Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell 2020;181:1329–45.
- Bakir B , Chiarella AM , Pitarresi JR , Rustgi AK . EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol 2020;30:764–76.
- Gupta PB , Pastushenko I , Skibinski A , Blanpain C , Kuperwasser C . Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 2019;24:65–78.
- Lambert AW , Weinberg RA . Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer 2021;21:325–38.
- Lindner P , Paul S , Eckstein M , Hampel C , Muenzner JK , Erlenbach-Wuensch K et al . EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis 2020;11:147.
- Javaid S , Zhang J , Anderssen E , Black JC , Wittner BS , Tajima K et al . Dynamic chromatin modification sustains epithelial-mesenchymal transition following inducible expression of Snail-1. Cell Rep 2013;5:1679–89.
- Serrano-Gomez SJ , Maziveyi M , Alahari SK . Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 2016;15:18.
- Skrypek N , Goossens S , De Smedt E , Vandamme N , Berx G . Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet TIG 2017;33:943–59.
- Li L , Hanahan D . Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 2013;153:86–100.
- Li L , Zeng Q , Bhutkar A , Galván JA , Karamitopoulou E , Noordermeer D et al . GKAP acts as a genetic modulator of NMDAR signaling to govern invasive tumor growth. Cancer Cell 2018;33:736–51.
- Mohammadi H , Sahai E . Mechanisms and impact of altered tumour mechanics. Nat Cell Biol 2018;20:766–74.
- Odenthal J , Takes R , Friedl P . Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis 2016;37:1117–28.
- Torres CM , Biran A , Burney MJ , Patel H , Henser-Brownhill T , Cohen A-HS et al . The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 2016;353:aaf1644.
- Puram SV , Tirosh I , Parikh AS , Patel AP , Yizhak K , Gillespie S et al . Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171:1611–24.
- Kinker GS , Greenwald AC , Tal R , Orlova Z , Cuoco MS , McFarland JM et al . Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 2020;52:1208–18.
- Murtha M , Esteller M . Extraordinary cancer epigenomics: thinking outside the classical coding and promoter box. Trends Cancer 2016;2:572–84.
- Nebbioso A , Tambaro FP , Dell’Aversana C , Altucci L . Cancer epigenetics: moving forward. PLoS Genet 2018;14:e1007362.
- Tavernari D , Battistello E , Dheilly E , Petruzzella AS , Mina M , Sordet-Dessimoz J et al . Non-genetic evolution drives lung adenocarcinoma spatial heterogeneity and progression. Cancer Discov 2021;11:1490–507.
- Heyn H , Vidal E , Ferreira HJ , Vizoso M , Sayols S , Gomez A et al . Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 2016;17:11.
- Saghafinia S , Mina M , Riggi N , Hanahan D , Ciriello G . Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 2018;25:1066–80.
- Audia JE , Campbell RM . Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8:a019521.
- Corces MR , Granja JM , Shams S , Louie BH , Seoane JA , Zhou W et al . The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898.
- Esteve-Puig R , Bueno-Costa A , Esteller M . Writers, readers and erasers of RNA modifications in cancer. Cancer Lett 2020;474:127–37.
- Janin M , Coll-SanMartin L , Esteller M . Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer 2020;19:70.
- Hanahan D , Coussens LM . Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–22.
- Lu Z , Zou J , Li S , Topper MJ , Tao Y , Zhang H et al . Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020;579:284–90.
- Thomas S , Izard J , Walsh E , Batich K , Chongsathidkiet P , Clarke G et al . The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res 2017;77:1783–812.
- Dzutsev A , Badger JH , Perez-Chanona E , Roy S , Salcedo R , Smith CK et al . Microbes and cancer. Annu Rev Immunol 2017;35:199–228.
- Helmink BA , Khan MAW , Hermann A , Gopalakrishnan V , Wargo JA . The microbiome, cancer, and cancer therapy. Nat Med 2019;25:377–88.
- Sears CL , Garrett WS . Microbes, microbiota, and colon cancer. Cell Host Microbe 2014;15:317–28.
- Pleguezuelos-Manzano C , Puschhof J , Rosendahl Huber A , van Hoeck A , Wood HM , Nomburg J et al . Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020;580:269–73.
- Okumura S , Konishi Y , Narukawa M , Sugiura Y , Yoshimoto S , Arai Y et al . Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat Commun 2021;12:5674.
- Salvi PS , Cowles RA . Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in homeostasis and disease. Cells 2021;10:1775.
- Fessler J , Matson V , Gajewski TF . Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 2019;7:108.
- Gopalakrishnan V , Helmink BA , Spencer CN , Reuben A , Wargo JA . The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 2018;33:570–80.
- Zitvogel L , Ma Y , Raoult D , Kroemer G , Gajewski TF . The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–70.
- Baruch EN , Youngster I , Ben-Betzalel G , Ortenberg R , Lahat A , Katz L et al . Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021;371:602–9.
- Davar D , Dzutsev AK , McCulloch JA , Rodrigues RR , Chauvin J-M , Morrison RM et al . Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021;371:595–602.
- Griffin ME , Espinosa J , Becker JL , Luo J-D , Carroll TS , Jha JK et al . Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 2021;373:1040–6.
- Mager LF , Burkhard R , Pett N , Cooke NCA , Brown K , Ramay H et al . Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369:1481–9.
- Ansaldo E , Belkaid Y . How microbiota improve immunotherapy. Science 2021;373:966–7.
- Ansaldo E , Farley TK , Belkaid Y . Control of immunity by the microbiota. Annu Rev Immunol 2021;39:449–79.
- Zhang Q , Ma C , Duan Y , Heinrich B , Rosato U , Diggs LP et al . Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov 2021;11:1248–67.
- Ding T , Schloss PD . Dynamics and associations of microbial community types across the human body. Nature 2014;509:357–60.
- Byrd AL , Liu M , Fujimura KE , Lyalina S , Nagarkar DR , Charbit B et al . Gut microbiome stability and dynamics in healthy donors and patients with non-gastrointestinal cancers. J Exp Med 2021;218:e20200606.
- Healy CM , Moran GP . The microbiome and oral cancer: more questions than answers. Oral Oncol 2019;89:30-3.
- Swaney MH , Kalan LR . Living in your skin: microbes, molecules and mechanisms. Infect Immun 2021;89:e00695–20.
- Willis JR , Gabaldón T . The human oral microbiome in health and disease: from sequences to ecosystems. Microorganisms 2020;8:308.
- Xu J , Peng J-J , Yang W , Fu K , Zhang Y . Vaginal microbiomes and ovarian cancer: a review. Am J Cancer Res 2020;10:743–56.
- Nejman D , Livyatan I , Fuks G , Gavert N , Zwang Y , Geller LT et al . The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80.
- Jin C , Lagoudas GK , Zhao C , Bullman S , Bhutkar A , Hu B et al . Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019;176:998–1013.
- Pushalkar S , Hundeyin M , Daley D , Zambirinis CP , Kurz E , Mishra A et al . The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 2018;8:403–16.
- McAllister F , Khan MAW , Helmink B , Wargo JA . The tumor microbiome in pancreatic cancer: bacteria and beyond. Cancer Cell 2019;36:577–9.
- Kadosh E , Snir-Alkalay I , Venkatachalam A , May S , Lasry A , Elyada E et al . The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020;586:133–8.
- Birch J , Gil J . Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34:1565–76.
- Faget DV , Ren Q , Stewart SA . Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 2019;19:439–53.
- Gorgoulis V , Adams PD , Alimonti A , Bennett DC , Bischof O , Bishop C et al . Cellular senescence: defining a path forward. Cell 2019;179:813–27.
- He S , Sharpless NE . Senescence in health and disease. Cell 2017;169:1000–11.
- Kowald A , Passos JF , Kirkwood TBL . On the evolution of cellular senescence. Aging Cell 2020;19:e13270.
- Lee S , Schmitt CA . The dynamic nature of senescence in cancer. Nat Cell Biol 2019;21:94–101.
- Wang B , Kohli J , Demaria M . Senescent cells in cancer therapy: friends or foes? Trends Cancer 2020;6:838–57.
- Baker DJ , Childs BG , Durik M , Wijers ME , Sieben CJ , Zhong J et al . Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–9.
- Ruhland MK , Loza AJ , Capietto A-H , Luo X , Knolhoff BL , Flanagan KC et al . Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 2016;7:11762.
- Hwang HJ , Lee Y-R , Kang D , Lee HC , Seo HR , Ryu J-K et al . Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett 2020;490:100–10.
- Wang D , Xiao F , Feng Z , Li M , Kong L , Huang L et al . Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res 2020;22:103.
- Amor C , Feucht J , Leibold J , Ho Y-J , Zhu C , Alonso-Curbelo D et al . Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020;583:127–32.
- Aster JC , Pear WS , Blacklow SC . The varied roles of notch in cancer. Annu Rev Pathol 2017;12:245–75.
- Kuczynski EA , Vermeulen PB , Pezzella F , Kerbel RS , Reynolds AR . Vessel co-option in cancer. Nat Rev Clin Oncol 2019;16:469–93.
Google ScholarCrossref