 Suche
Suche
 Mein Konto
Mein Konto
Kännetecken för cancer: Nya dimensioner

Förord The Hallmarks of Cancer Conceptualization är ett heuristiskt verktyg för att destillera den enorma komplexiteten hos cancerfenotyper och genotyper till en preliminär uppsättning underliggande principer. I takt med att kunskapen om cancermekanismer har utvecklats har andra aspekter av sjukdomen dykt upp som potentiella förbättringar. Detta väcker utsikterna att fenotypisk plasticitet och oordnad differentiering är en distinkt karakteristisk förmåga, och att icke-mutationell epigenetisk omprogrammering och polymorfa mikrobiomer båda representerar karakteristiska möjliggörande egenskaper som underlättar förvärvet av karakteristiska förmågor. Vidare kan åldrande celler av olika ursprung läggas till listan över funktionellt viktiga celltyper i tumörmikromiljön. Det betyder att cancer är skrämmande i...

Kännetecken för cancer: Nya dimensioner
Förord
The Hallmarks of Cancer Conceptualization är ett heuristiskt verktyg för att destillera den enorma komplexiteten hos cancerfenotyper och genotyper till en preliminär uppsättning underliggande principer. I takt med att kunskapen om cancermekanismer har utvecklats har andra aspekter av sjukdomen dykt upp som potentiella förbättringar. Detta väcker utsikterna att fenotypisk plasticitet och oordnad differentiering är en distinkt karakteristisk förmåga, och att icke-mutationell epigenetisk omprogrammering och polymorfa mikrobiomer båda representerar karakteristiska möjliggörande egenskaper som underlättar förvärvet av karakteristiska förmågor. Vidare kan åldrande celler av olika ursprung läggas till listan över funktionellt viktiga celltyper i tumörmikromiljön.
Menande
Cancer är skrämmande i bredden och omfattningen av dess mångfald, vilket inkluderar genetik, cell- och vävnadsbiologi, patologi och respons på terapi. Allt kraftfullare experimentella och beräkningsverktyg och tekniker tillhandahåller en lavin av "big data" om de otaliga sjukdomsmanifestationer som cancer omfattar. Det integrerande konceptet som förkroppsligas i cancers kännetecken hjälper till att destillera denna komplexitet till en allt mer logisk vetenskap, och de preliminära nya dimensionerna som presenteras i detta perspektiv kan ge mervärde till denna strävan att bättre förstå mekanismerna för karcinogenes och malign progression och tillämpa denna kunskap på cancermedicin.
introduktion
Kräftens kännetecken har föreslagits som en uppsättning funktionella förmågor som mänskliga celler förvärvar när de går från normalitet till neoplastiska tillväxttillstånd, mer specifikt förmågor som är avgörande för deras förmåga att bilda maligna tumörer. I dessa artiklar ( 1, 2 ), listade Bob Weinberg och jag vad vi föreställde oss som gemensamma drag som förenar alla typer av cancerceller på nivån av cellulär fenotyp. Avsikten var att tillhandahålla ett konceptuellt ramverk som skulle tillåta de komplexa fenotyperna av olika mänskliga tumörtyper och varianter att rationaliseras i förhållande till en gemensam uppsättning av underliggande cellulära parametrar. Inledningsvis föreställde vi oss att sex olika varumärkesfunktioner skulle kompletteras och utökades senare till åtta.
Denna formulering påverkades av insikten att cancer hos människa utvecklas som produkter av flerstegsprocesser och att förvärvet av dessa funktionella förmågor på något sätt kan tillskrivas de distinkta stegen av tumörpatogenes. Mångfalden av malign patogenes, som omfattar flera tumörtyper och en ökande mängd subtyper, involverar olika avvikelser (och därmed förvärvade förmågor och egenskaper) som är resultatet av vävnadsspecifika barriärer som nödvändigtvis förbigås under vissa tumörbildningsvägar. Även om vi inser att sådana specialiserade mekanismer kan vara till hjälp, har vi begränsat beteckningen av kännetecknen till parametrar som har en bred inverkan över hela spektrumet av mänskliga cancerformer.
De åtta kännetecknen inkluderar för närvarande (Fig.1, till vänster) de förvärvade förmågorna att upprätthålla proliferativ signalering, undvika tillväxtdämpare, motstå celldöd, möjliggöra replikativ odödlighet, inducera/åtkomst till kärl, aktivera invasion och metastasering, omprogrammera cellulär metabolism och undvika förstörelse av immunsystemet. I den senaste utarbetandet av detta koncept (2) avgränsades avreglering av cellulär metabolism och undvikande av förstörelse av immunsystemet som "framväxande kännetecken", men nu, elva år senare, är det uppenbart att de, i likhet med de ursprungliga sex, kan betraktas som kärnkännetecken för cancer och ingår som sådana i den aktuella berättelsen (Fig. 1, vänster).
Figur 1
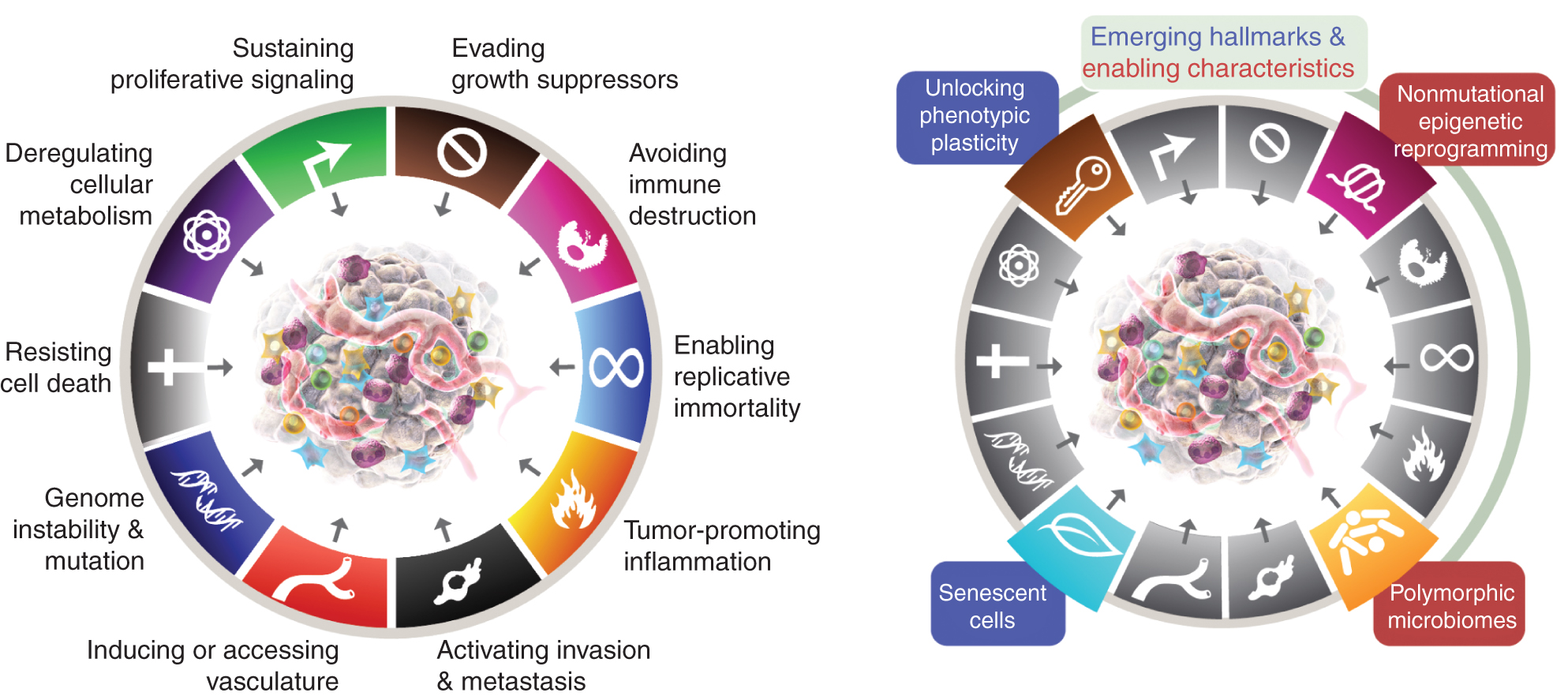
Kräftans kännetecken förkroppsligar för närvarande åtta distinkta förmågor och två stödjande egenskaper. Förutom de sex förvärvade förmågorna - kännetecken för cancer - som föreslogs 2000 (1), har de två preliminära "emergenta kännetecknen" som introducerades 2011 (2) - cellulär energi (nu mer allmänt kallad "omprogrammering av cellulär metabolism") och "undvika immunförstöring" - ansetts vara tillräckligt validerade för att uppsättningen är tillräckligt validerade.
Med tanke på det växande erkännandet att tumörer kan vaskulariseras på ett adekvat sätt, antingen genom att slå på angiogenes eller genom att samordna normal vävnadskärlsystem (128), definieras detta kännetecken också bredare som förmågan att inducera eller på annat sätt få tillgång till kärlsystem som stöder tumörtillväxt främst genom invasion och metastasering.
2011 års uppföljare inkluderade också "tumörfrämjande inflammation" som en andra möjliggörande egenskap, som kompletterar den övergripande "genominstabiliteten och mutationen", som tillsammans var fundamentalt involverade i att aktivera de åtta signaturförmågorna (funktionella) som krävs för tumörtillväxt och -progression. Det är sant att den här recensionen inkluderar ytterligare föreslagna nya kännetecken och möjliggörande funktioner, inklusive "låsa upp fenotypisk plasticitet", "icke-mutationell epigenetisk omprogrammering", "polymorfa mikrobiomer" och "åldrande celler." Varumärkena för cancergrafiken antogs från Hanahan och Weinberg (2).
Som vi noterade vid den tiden, kan dessa särskiljande särdrag ensamma inte ta itu med komplexiteten av cancerpatogenes, dvs. de exakta molekylära och cellulära mekanismerna som gör det möjligt för de utvecklande preneoplastiska cellerna att utveckla och förvärva dessa avvikande fenotypiska förmågor under förloppet av tumörbildning och malign progression.
Följaktligen har vi lagt till ett annat koncept till diskussionen som presenteras som "möjliggörande egenskaper", konsekvenser av det avvikande tillståndet hos neoplasman som tillhandahåller medel för att cancerceller och tumörer kan förvärva dessa funktionella egenskaper. Som sådan återspeglas de möjliggörande egenskaperna i molekylära och cellulära mekanismer genom vilka kännetecken förvärvas, snarare än i de ovanstående åtta färdigheterna själva. Dessa två aktiveringsprocesser var genominstabilitet och tumörfrämjande inflammation.
Vi insåg vidare att tumörmikromiljön (TME), definierad häri som sammansatt av heterogena och interaktiva populationer av cancerceller och cancerstamceller tillsammans med en mängd olika rekryterade stromacelltyper - det transformerade parenkymet och associerade stroma - nu är allmänt uppskattad för att spela en viktig roll i tumörbildning och malign progression.
Med tanke på det pågående intresset för dessa formuleringar och vår fortsatta avsikt att uppmuntra pågående diskussion och förfining av Hallmarks-schemat, är det lämpligt att överväga en ofta ställd fråga: Finns det ytterligare funktioner i denna konceptuella modell som skulle kunna införlivas, med hänsyn till behovet av att säkerställa detta? att de är brett tillämpliga över hela spektrumet av mänskliga cancerformer? Följaktligen presenterar jag flera potentiella nya kännetecken och möjliggörande funktioner som i sinom tid skulle kunna integreras som kärnkomponenter i kännetecknen för cancerkonceptualisering.
Dessa parametrar är att "låsa upp fenotypisk plasticitet", "icke-mutationell epigenetisk omprogrammering", "polymorfa mikrobiomer" och "åldrande celler" (Fig. 1, höger). Viktigt är att de exempel som presenteras till stöd för dessa teser är illustrativa men inte på något sätt heltäckande, eftersom det finns en växande och allt mer övertygande mängd publicerade bevis som stödjer varje vinjett.
Tapping fenotypisk plasticitet
Under organogenes åtföljs utvecklingen, bestämningen och organiseringen av celler till vävnader för att utföra homeostatiska funktioner av terminal differentiering, med stamceller som upphör att växa, ibland irreversibelt, eftersom dessa processer kulminerar. Som sådant är slutresultatet av cellulär differentiering, i de flesta fall, antiproliferativt, vilket bildar en tydlig barriär för fortsatt proliferation som är nödvändig för neoplasi.
Det finns allt fler bevis för att låsning av den normalt begränsade förmågan för fenotypisk plasticitet att kringgå eller undkomma tillståndet av terminal differentiering är en kritisk komponent i cancerpatogenes (3). Denna plasticitet kan verka i flera manifestationer (fig. 2). Sålunda kan begynnande cancerceller som härstammar från en normal cell som har utvecklats längs en väg som närmar sig eller antar ett helt differentierat tillstånd vända kursen genom att dedifferentiera tillbaka till progenitorliknande celltillstånd.
Omvänt kan neoplastiska celler som härrör från en progenitorcell som är avsedd att följa en väg som leder till terminal differentiering kortsluta processen och bibehålla de expanderande cancercellerna i ett delvis differentierat, progenitorliknande tillstånd. Alternativt kan transdifferentiering inträffa där celler som ursprungligen engagerade sig i en differentieringsväg byter till ett helt annat utvecklingsprogram och därigenom förvärvar vävnadsspecifika egenskaper som inte var förutbestämda av deras normala ursprungsceller.
Följande exempel stödjer argumentet att olika former av cellulär plasticitet avslöjar fenotypisk plasticitet. Till vänster är fenotypisk plasticitet utan tvekan en förvärvad karakteristisk förmåga som möjliggör olika störningar av celldifferentiering, inklusive (i) dedifferentiering från mogna till progenitortillstånd, (ii) avstannade (terminala) differentiering från progenitorcellstillstånd och (iii) transdifferentiering till andra cellinjer. Tre framträdande sätt för nedsatt differentiering som är integrerade i cancerpatogenes visas till höger.
Genom att differentiellt pervertera den normala differentieringen av progenitorceller till mogna celler i utvecklingslinjer, underlättas tumörbildning och malign progression som härrör från celler av ursprung i sådana vägar. Varumärkena för cancergrafiken antogs från Hanahan och Weinberg (2).
Figur 2

Avdifferentiering
Koloncancer är ett exempel på försämrad differentiering, eftersom det finns ett teleologiskt behov för begynnande cancerceller att fly transportbandet av terminal differentiering och exfoliering, vilket i princip skulle kunna ske genom dedifferentiering av kolonepitelceller som ännu inte terminalt differentierat eller genom avstannad differentiering av dessa cellföräldrar till olika stamceller som ger olika stamceller. celler. Både differentierade celler och stamceller har varit inblandade som ursprungsceller för tjocktarmscancer ( 4 – 6 ).
Två utvecklingsmässiga transkriptionsfaktorer (TF), homeobox-proteinet HOXA5 och SMAD4, det senare involverat i BMP-signalering, uttrycks starkt i differentierande kolonepitelceller och går vanligtvis förlorade i avancerade kolonkarcinom, som karakteristiskt uttrycker markörer för stam- och progenitorceller. Funktionella störningar i musmodeller har visat att påtvingat uttryck av HOXA5 i tjocktarmscancerceller återställer differentieringsmarkörer, undertrycker stamcellsfenotyper och försämrar invasion och metastaser, vilket ger en motivering för dess karakteristiska nedreglering (7, 8).
Däremot framtvingar SMAD4 både differentiering och undertryckande av proliferation driven av onkogen WNT-signalering, vilket avslöjas av den konstruerade förlusten av SMAD4-uttryck, vilket ger en förklaring till dess förlust av uttryck för att tillåta dedifferentiering och därefter WNT-driven hyperproliferation (5).
Noterbart är att förlusten av dessa två "differentieringsundertryckare" med den resulterande avdifferentieringen är associerad med förvärvet av andra känneteckenförmågor, såväl som andra känneteckenframkallande regulatorer, vilket komplicerar den strikta definitionen av detta provisoriska kännetecken som separerbart och oberoende.
En annan linje av bevis rör det undertryckta uttrycket av MITF-mästarregulatorn för melanocytdifferentiering, som verkar vara involverad i uppkomsten av aggressiva former av malignt melanom. Förlust av denna utvecklings-TF är associerad med reaktivering av neural crest progenitor-gener och nedreglering av gener som karakteriserar helt differentierade melanocyter. Återuppträdandet av generna för neurala krön indikerar att dessa celler återgår till det progenitortillstånd från vilket melanocyter uppstår utvecklingsmässigt.
Vidare etablerade en linjespårningsstudie av BRAF-inducerade melanom mogna pigmenterade melanocyter som ursprungsceller som genomgår dedifferentiering under tumörbildningsförloppet (9). Noterbart är att den mutanta BRAF-onkogenen, som finns i mer än hälften av kutana melanom, inducerar hyperproliferation, som föregår och kan därför separeras mekaniskt från den efterföljande dedifferentieringen som uppstår från nedreglering av MITF.
En annan studie implicerade funktionellt uppregleringen av utvecklings-TF ATF2, vars karakteristiska uttryck i mus- och humanmelanom indirekt undertrycker MITF1, samtidigt med malign progression av de följaktligen dedifferentierade melanomcellerna (10). Omvänt resulterar uttryck i melanom av mutanta former av ATF2 som inte kan undertrycka MITF i väldifferentierade melanom (11).
Dessutom har en nyligen genomförd studie (12) kopplat avdifferentiering av härstamning till malign progression av neoplasmer i pankreatiska öar till metastasbenägna karcinom; dessa neuroendokrina celler och härledda tumörer härrör från en utvecklingslinje som är skild från den som genererar det mycket större antalet närliggande celler som bildar exokrina och pankreas och de resulterande ductala adenocarcinomen.
Anmärkningsvärt nog har flerstegsdifferentieringsvägen från ö-stamceller till mogna β-celler karakteriserats grundligt (13). Jämförande transkriptomprofilering visar att adenomliknande ö-tumörer mest liknar omogna men differentierade insulinproducerande β-celler, medan de invasiva karcinomen liknar mest embryonala öcellsprekursorer. Progression till dåligt differentierade karcinom innebär ett initialt steg av dedifferentiering, som initialt inte innebär ökad proliferation eller minskad apoptos jämfört med väldifferentierade adenom, som båda tenderar att inträffa senare.
Det diskreta steget med dedifferentiering drivs således inte av observerbara förändringar i de karakteristiska särdragen för ihållande proliferation och resistens mot apoptos. Snarare är uppregleringen av ett miRNA som tidigare var inblandat i att specificera öarnas stamfadertillstånd en som nedregleras under terminal differentiering av β-celler, 12).
Blockerad differentiering
Medan ovanstående exempel illustrerar hur undertryckande av differentieringsfaktoruttryck kan underlätta tumörbildning genom att tillåta bättre differentierade celler att dedifferentiera till stamceller, kan i andra fall ofullständigt differentierade stamceller drabbas av regulatoriska förändringar som aktivt blockerar deras fortsatta progression till helt differentierade, typiskt icke-proliferativa tillstånd.
Det har länge dokumenterats att akut promyelocytisk leukemi (APL) är ett resultat av en kromosomal translokation som sammansmälter PML-lokuset till genen som kodar för retinsyra α-kärnreceptorn (RARα). Myeloida progenitorceller som bär sådana translokationer är uppenbarligen oförmögna att fortsätta sin vanliga terminala differentiering till granulocyter, vilket resulterar i celler fångade i ett proliferativt, promyelocytliknande progenitorstadium (14).
Bevis på konceptet för detta schema kommer från behandlingen av odlade APL-celler, musmodeller av sjukdomen och drabbade patienter med retinsyra, liganden av RARα; Denna terapeutiska behandling får de neoplastiska APL-cellerna att differentiera till till synes mogna, icke-prolifererande granulocyter, och kortsluter därigenom deras progressiva proliferativa expansion (14–16).
En variant på detta tema gäller en annan form av akut myeloid leukemi, denna som bär på t(8;21)-translokationen som producerar AML1-ETO-fusionsproteinet. Detta protein ensamt kan transformera myeloida progenitorer, åtminstone delvis genom att blockera deras differentiering. Terapeutisk intervention i musmodeller och patienter med en farmakologisk hämmare av ett kromatinmodifierande histondeacetylas (HDAC) får myeloida leukemiceller att återuppta differentiering till celler med en mer mogen myeloid cellmorfologi. Till denna reaktion hör en minskning av proliferativ förmåga, vilket försämrar utvecklingen av denna leukemi (17, 18).
Ett tredje exempel på melanom involverar en utvecklings-TF, SOX10, som normalt nedregleras under melanocytdifferentiering. Vinst och förlust av funktionsstudier i en zebrafiskmodell av BRAF-inducerade melanom har visat att onormalt bibehållet uttryck av SOX10 blockerar differentieringen av neurala progenitorceller till melanocyter, vilket möjliggör bildandet av BRAF-drivna melanom (19).
Andra exempel på differentieringsmodulatorer inkluderar metaboliten alfa-ketoglutarat (αKG), en nödvändig kofaktor för ett antal kromatinmodifierande enzymer som har visat sig vara involverad i att stimulera vissa differentierade celltillstånd. Vid pankreascancer stimulerar tumörsuppressorn p53 produktionen av αKG och upprätthållandet av ett mer differentierat celltillstånd, medan en prototypisk förlust av p53-funktionen leder till en minskning av αKG-nivåerna och åtföljande dedifferentiering, vilket är associerat med malign progression (20).
I en form av levercancer resulterar mutation av en isocitratdehydrogenasgen (IDH1/2) inte i produktionen av differentieringsinducerande αKG, utan snarare en relaterad "oncometabolite", D-2-hydroxigluterat (D2HG), som har visat sig blockera hepatocytdifferentiering av leverprogenitorceller i leverns stamceller genom D2-regulatorer av G-celler som reglerar masterceller och aH-celler. och stillastående, HNF4a.
D2HG-medierad suppression av HNF4a-funktionen utlöser en proliferativ expansion av hepatocytprogenitorceller i levern, som blir mottagliga för onkogen transformation vid efterföljande mutationsaktivering av KRAS-onkogenen, som driver malign progression till kolangiokarcinom i levern (21). IDH1/2-mutanten och dess onkometabolit D2HG fungerar också i en mängd myeloida och andra solida tumörtyper, där D2HG hämmar αKG-beroende dioxygenaser som krävs för histon- och DNA-metyleringshändelser som medierar förändringar i kromatinstrukturen under differentiering av utvecklingslinje, och därigenom fryser uppkomsten av cancerceller i a2 progeni2-tillstånd (a2 progeni23).
Ett ytterligare, relaterat koncept är "förbikopplad differentiering", där partiellt eller odifferentierade progenitor-/stamceller lämnar cellcykeln och ligger vilande i skyddande nischer, med potential att återinitiera proliferativ expansion (24), men fortfarande med det selektiva trycket att störa sin programmerade differentiering på ett eller annat sätt.
Transdifferentiering
Begreppet transdifferentiering har länge erkänts av patologer i form av vävnadsmetaplasi, där celler av en viss differentierad fenotyp markant ändrar sin morfologi för att bli tydligt igenkännbara som element i en annan vävnad, ett framträdande exempel på vilket är Barretts matstrupe, där kronisk inflammation i den stratifierade stratifierade epitheliquishages transdifferentiering till ett enkelt kolumnärt epitel som är karakteristiskt för tarmen, vilket underlättar den efterföljande utvecklingen av adenokarcinom snarare än de skivepitelcancer som förväntas från detta skivepitel (3).
Nu avslöjar molekylära determinanter mekanismer för transdifferentiering i olika cancerformer, både för fall där grov vävnadsmetaplasi är uppenbar och andra där den är något mer subtil, som följande exempel illustrerar.
Ett informativt fall för transdifferentiering som en diskret händelse vid tumörbildning rör pankreatiskt duktalt adenokarcinom (PDAC), i vilket en av de involverade ursprungscellerna, pankreatisk acinarcell, kan transdifferentiera till en duktal cellfenotyp under initieringen av neoplastisk utveckling. Två TF: er - PTF1a och MIST1 - kontrollerar specifikationen och underhållet av det differentierade acinära celltillståndet i bukspottkörteln via deras uttryck i samband med självförsörjande "feed-forward" regulatoriska loopar (25).
Båda dessa TF: er nedregleras ofta under neoplastisk utveckling och malign progression av human och mus PDAC. Funktionella genetiska studier i möss och odlade humana PDAC-celler har visat att experimentellt påtvingat uttryck av PTF1a försämrar KRAS-inducerad transdifferentiering och proliferation och kan också tvinga omdifferentiering av redan neoplastiska celler till en vilande acinär cellfenotyp (26).
Omvänt utlöser suppression av PTF1a-uttryck acinar-to-duct-metaplasi, nämligen transdifferentiering, och sensibiliserar därigenom de kanalliknande cellerna för onkogen KRAS-transformation, vilket påskyndar den efterföljande utvecklingen av invasiv PDAC (27). På samma sätt blockerar påtvingat uttryck av MIST1 i KRAS-uttryckande pankreas transdifferentiering och försämrar initieringen av pankreatisk tumörbildning, vilket annars underlättas av bildandet av premaligna kanalliknande (PanIN) lesioner, medan genetisk deletion av MIST1 förbättrar deras bildning och initieringen av neoplastisk 2KRAS.
Förlust av antingen PTF1- eller MIST1-uttryck under tumörbildning är associerat med ökat uttryck av en annan utvecklingsregulatorisk TF, SOX9, som normalt är effektiv i ductal cellspecifikation (27, 28). Påtvingad uppreglering av SOX9, och därigenom undviker behovet av nedreglering av PTF1a, och MIST1 har också visat sig stimulera transdifferentieringen av acinära celler till en duktal cellfenotyp som är känslig för KRAS-inducerad neoplasi (29), vilket implicerar SOX9 som en nyckelfunktionell effektor av deras nedreglering av humanPDAC.
Således kan tre TF: er som reglerar pankreatisk differentiering ändras på olika sätt för att inducera ett transdifferentierat tillstånd som, i samband med mutationsaktivering av KRAS, underlättar onkogen transformation och initieringen av tumörbildning och malign progression.
Ytterligare medlemmar av SOX-familjen av kromatinassocierade regulatoriska faktorer är å ena sidan till stor del associerade med både cellödespecifikation och linjebyte under utveckling (30) och å andra sidan med flera tumörassocierade fenotyper (31). Ett annat framträdande exempel på SOX-medierad transdifferentiering involverar en mekanism för terapeutisk resistens vid prostatacancer.
I det här fallet är förlust av RB- och p53-tumörsuppressorerna - vars frånvaro är karakteristisk för neuroendokrina tumörer - som svar på antiandrogenterapi nödvändig men inte tillräcklig för den vanligen observerade transformationen av väldifferentierade prostatacancerceller till karcinomceller som har invaderat differentieringslinjen med molekylära och histologiska särdrag i molekylär- och histologiska celler. androgenreceptor. Förutom förlust av RB och p53 kräver förvärvad resistens mot antiandrogenterapi uppreglerat uttryck av SOX2, en utvecklingsregulatorisk gen, som har visat sig hjälpa till att inducera transdifferentiering av de terapikänsliga adenokarcinomcellerna till derivat som är i ett neuroendokrint celltillstånd som är refraktärt mot terapi (32).
Ett tredje exempel visar också transdifferentiering som en strategi som används av karcinomceller för att undvika eliminering genom härstamningsspecifik terapi, i detta fall med basalcellscancer (BCC) i huden som behandlats med en farmakologisk hämmare av Hedgehog-Smoothened (HH/SMO) onkogen väg som är känd för att driva den neoplastiska tillväxten (33).
Läkemedelsresistenta cancerceller byter till en utvecklingsrelaterad men distinkt celltyp via breda epigenetiska förändringar i specifika kromatindomäner och förändrad tillgänglighet för två superförstärkare. Det nyförvärvade fenotypiska tillståndet hos BCC-celler tillåter dem att upprätthålla uttrycket av den onkogena WNT-signalvägen, vilket i sin tur ger oberoende från den läkemedelsundertryckta HH/SMO-signalvägen (34).
Som förväntat från denna transdifferentiering, skiftar transkriptomet av cancercellerna från en gensignatur som återspeglar den involverade ursprungscellen för BCC, nämligen hårsäckens utbuktande stamceller, till en signatur som indikerar de basala stamcellerna som befolkar BCC interfollikulär epidermis. Sådan transdifferentiering för att möjliggöra läkemedelsresistens dokumenteras alltmer vid olika former av cancer (35).
Plasticitet i utvecklingslinjen verkar också vara utbredd i de viktigaste subtyperna av lungkarcinom, d.v.s. vid neuroendokrina karcinom [småcellig lungcancer (SCLC)] och adenokarcinom + skivepitelcancer [kollektiv icke-småcellig lungcancer (NSCLC)]. Enkelcellig RNA-sekvensering har avslöjat anmärkningsvärt dynamisk och heterogen omvandling mellan dessa subtyper, såväl som markanta variationer däri, under stadierna av lungtumörbildning, efterföljande malign progression och svar på terapi (36-38).
Därför, snarare än den enkla konceptualiseringen av en ren klonal växling från en släktlinje till en annan, målar dessa studier en mycket mer komplex bild av dynamiskt interkonverterande subpopulationer av cancerceller som uppvisar drag av flera utvecklingslinjer och differentieringsstadier, en nykter insikt i detta avseende för linjebaserad terapeutisk inriktning av mänsklig lungcancer. Regulatoriska bestämningsfaktorer för denna dynamiska fenotypiska plasticitet börjar identifieras (37, 39, 40).
Sammanfattning
De tre klasserna av mekanismer som beskrivs ovan lyfter fram selektiva regulatorer av cellulär plasticitet som är - åtminstone delvis - separerbara från onkogena kärndrivrutiner och andra distinkta förmågor. Utöver dessa exempel finns det en betydande mängd bevis som kopplar många former av cancer till nedsatt differentiering, vilket åtföljs av förvärvet av transkriptomsignaturer och andra fenotyper - till exempel histologisk morfologi - som är associerade med progenitor- eller stamcellsstadier observerade i motsvarande normala vävnader. ursprung eller i andra mer avlägset besläktade celltyper och linjer (41 – 43).
Som sådana tycks dessa tre underklasser av fenotypisk plasticitet – avdifferentiering av mogna celler tillbaka till progenitortillstånd, avstannad differentiering för att frysa utvecklande celler i progenitor-/stamcellstillstånd och transdifferentiering till alternativa celllinjer – vara effektiva i flera cancertyper under primär tumörgenes, malignt framsteg och/eller terapi.
Det finns dock två konceptuella överväganden. För det första är dedifferentiering och avstängd differentiering sannolikt sammanflätade, eftersom de inte går att särskilja i många tumörtyper där ursprungscellen - differentierad cell eller progenitor / stamcell - är antingen okänd eller alternativt involverad. För det andra är förvärvet eller upprätthållandet av progenitorcellsfenotyper och förlusten av differentierade egenskaper, i de flesta fall, en felaktig återspegling av det normala utvecklingsstadiet, nedsänkt i en miljö av andra karakteristiska förändringar i cancercellen som inte finns i naturligt utvecklande celler.
Dessutom involverar en annan form av fenotypisk plasticitet cellulär senescens, diskuterad mer allmänt nedan, varvid cancerceller som induceras att genomgå till synes irreversibelt åldrande istället kan fly och fortsätta proliferativ expansion (44). Slutligen, som med andra distinkta förmågor, är cellulär plasticitet inte en ny uppfinning eller avvikelse av cancerceller, utan snarare korruptionen av latenta men aktiverbara förmågor som olika normala celler använder för att stödja homeostas, reparation och regenerering (45).
Sammantaget uppmuntrar dessa illustrativa exempel att överväga att upplåsning av cellulär plasticitet för att möjliggöra olika former av störd differentiering representerar en distinkt distinkt förmåga som skiljer sig i reglering och cellulär fenotyp från de välvaliderade kärnkännetecknen för cancer (Fig. 2).
Epigenetisk omprogrammering utan mutation
Den möjliggörande egenskapen hos genom (DNA) instabilitet och mutation är en grundläggande komponent i cancerutveckling och patogenes. För närvarande katalogiserar flera internationella konsortier mutationer i genomet av mänskliga cancerceller, i praktiskt taget alla typer av human cancer, i olika stadier av malign progression, inklusive metastaserande lesioner, och under utvecklingen av adaptiv terapiresistens. Ett resultat är det nu utbredda erkännandet att mutationer i gener som organiserar, modulerar och upprätthåller kromatinarkitektur och därigenom reglerar genuttryck globalt i allt högre grad upptäcks och funktionellt kopplas till canceregenskaper (46-48).
Vidare finns det argument för en annan till synes oberoende form av genomomprogrammering som involverar rent epigenetiskt reglerade förändringar i genuttryck, en som skulle kunna kallas "icke-mutationell epigenetisk omprogrammering" (Fig. 3). Faktum är att tesen om mutationslös cancerevolution och rent epigenetisk programmering av karakteristiska cancerfenotyper togs upp för nästan ett decennium sedan (49) och diskuteras alltmer (46, 50–52).
Figur 3

I likhet med vad som sker under embryogenes och vävnadsdifferentiering och homeostas, tyder ackumulerande bevis på att instrumentella genreglerande kretsar och nätverk i tumörer kan kontrolleras av en uppsjö av korrupta och co-opterade mekanismer som är oberoende av genominstabilitet och genmutation. Varumärkena för cancergrafiken antogs från Hanahan och Weinberg (2).
Naturligtvis är begreppet icke-mutationell epigenetisk reglering av genuttryck väl etablerad som den centrala mekanismen som förmedlar embryonal utveckling, differentiering och organogenes (53-55). Hos vuxna, till exempel, involverar långtidsminnet förändringar i gen- och histonmodifiering, i kromatinstrukturen och i avfyrningen av genuttrycksomkopplare, som bibehålls stabilt över tiden av positiva och negativa återkopplingsslingor (56, 57). Ökande bevis stöder tanken att analoga epigenetiska förändringar kan bidra till förvärvet av karakteristiska förmågor under tumörutveckling och malign progression. För att stödja denna hypotes presenteras några exempel nedan.
Mikromiljömekanismer för epigenetisk omprogrammering
Om inte genom enbart onkogena mutationer, hur omprogrammeras cancercellens genom? En växande mängd bevis tyder på att de avvikande fysiska egenskaperna hos tumörmikromiljön kan orsaka breda förändringar i epigenomet, av vilka förändringar som är fördelaktiga för fenotypiskt urval av egenskaper kan leda till klonal utväxt av cancerceller med förbättrad kondition för proliferativ expansion.
Ett vanligt kännetecken för tumörer (eller regioner i tumörer) är hypoxi som ett resultat av otillräcklig vaskularisering. Hypoxi minskar till exempel aktiviteten hos TET-demetylaser, vilket leder till betydande förändringar i metylomen, särskilt hypermetylering (58). Otillräcklig vaskularisering kommer sannolikt också att begränsa biotillgängligheten av kritiska blodburna näringsämnen, och näringsbrist har till exempel visat sig förändra translationskontrollen och följaktligen öka den maligna fenotypen av bröstcancerceller (59).
Ett övertygande exempel på hypoximedierad epigenetisk reglering är en form av det alltid dödliga pediatriska ependymomet. Liksom många embryonala och pediatriska tumörer saknar denna form återkommande mutationer, särskilt en brist på förarmutationer i onkogener och tumörsuppressorer. Snarare har den onormala tillväxten av dessa cancerceller visat sig kontrolleras av ett hypoxiinducerat genreglerande program (60, 61). Anmärkningsvärt är att den förmodade ursprungscellen för denna cancer finns i ett hypoxiskt fack och sannolikt sensibiliserar celler i den för att initiera tumörbildning genom ännu okända kofaktorer.
Ett annat övertygande bevis för mikromiljömedierad epigenetisk reglering gäller cancercellers invasiva tillväxtförmåga. Ett klassiskt exempel är den reversibla induktionen av invasivitet hos cancerceller vid kanterna av många solida tumörer, orkestrerad av det utvecklingsmässiga regulatoriska programmet känt som epitel-till-mesenkymal övergång (EMT; refs. 62–64). Särskilt har en masterregulator för EMT, ZEB1, nyligen visat sig inducera uttrycket av ett histonmetyltransferas, SETD1B, som i sin tur upprätthåller ZEB1-uttryck i en positiv återkopplingsslinga som upprätthåller det (invasiva) regulatoriska tillståndet för EMT (65).
En tidigare studie dokumenterade på liknande sätt att induktion av EMT genom uppreglerat uttryck av en relaterad TF, SNAIL1, orsakade markanta förändringar i kromatinlandskapet som ett resultat av induktionen av ett antal kromatinmodifierare vars aktivitet visade sig vara nödvändig för att upprätthålla det fenotypiska tillståndet (66). Vidare kan ett antal tillstånd och faktorer som upplevs av cancerceller vid kanterna av tumörer, inklusive hypoxi och cytokiner som utsöndras av stromaceller, uppenbarligen inducera EMT och därmed invasivitet (67, 68).
Ett slående exempel på programmering av invasivitet av mikromiljön, som förmodligen inte är relaterad till EMT-programmet, involverar autokrin aktivering av en neuronal signalkrets som involverar utsöndrat glutamat och dess receptor NMDAR (69, 70). Anmärkningsvärt nog har den prototypiska stelheten hos många solida tumörer, förkroppsligad i omfattande förändringar av den extracellulära matrisen (ECM) som omsluter cellerna i dem, djupgående konsekvenser för cancercellernas invasiva och andra fenotypiska egenskaper.
Jämfört med den normala vävnads-ECM från vilken tumörer uppstår, kännetecknas tumör-ECM typiskt av ökad tvärbindning och densitet, enzymatiska modifieringar och förändrad molekylär sammansättning som kollektivt orkestrerar, delvis genom integrinreceptorer för ECM-motiv, stelhetsinducerad signalering och genuttrycksnätverk som inducerar invasiva egenskaper (7).
Utöver sådana regleringsmekanismer som tillhandahålls av den fysiska tumörmikromiljön, kan parakrin signalering, innefattande lösliga faktorer som frigörs i den extracellulära miljön av de olika celltyperna som befolkar solida tumörer, också bidra till induktionen av flera morfologiskt distinkta invasiva tillväxtprogram (72), av vilka endast ett – benämnts ovan benämnts ”epikement” – ovan regleringsmekanism för EMT.
Epigenetisk regulatorisk heterogenitet
En växande kunskapsbas ökar förståelsen för betydelsen av intratumoral heterogenitet för att generera den fenotypiska mångfalden där de mest lämpliga cellerna för proliferativ expansion och invasion växer ur sina bröder och därför väljs ut för malign progression. Visst, en aspekt av denna fenotypiska heterogenitet beror på kronisk eller episodisk genomisk instabilitet och resulterande genetisk heterogenitet i cellerna som befolkar en tumör.
Dessutom blir det allt tydligare att icke-mutationsbaserad epigenetisk heterogenitet kan existera. Ett framträdande exempel är linkerhistonen H1.0, som är dynamiskt uttryckt och undertryckt i subpopulationer av cancerceller inom en rad tumörtyper, med åtföljande sekvestrering eller tillgänglighet av megabasstora domäner [73]. Noterbart visade sig populationen av cancerceller med undertryckt H1.0 uppvisa stamliknande egenskaper, förbättrad tumörinitierande förmåga och ett samband med dålig prognos hos patienter.
Ett annat exempel på epigenetiskt reglerad plasticitet har beskrivits i humana orala skivepitelcancer (SCC), där cancerceller vid de invasiva marginalerna antar ett partiellt EMT-tillstånd (p-EMT) som saknar de tidigare nämnda mesenkymala TF:erna men uttrycker andra EMT-definierande gener som inte uttrycks i den centrala kärnan av tumören (74).
p-EMT-cellerna representerar uppenbarligen inte klonal kompartmentalisering av mutationsförändrade celler: kulturer av primära tumörhärledda cancerceller innehåller dynamiska blandningar av både p-EMT hi- och p-EMT-lo-celler och när p-EMT hi/lo-celler FACS-renades och odlades, återgick båda till blandade populationer av p-EMT-lohi och p-EMT-dagar. Även om parakrina signaler från det intilliggande stroma kan anses vara deterministiska för p-EMT hi-tillståndet, argumenterar den stabila närvaron och regenereringen av de två epigenetiska tillstånden i kulturen för en cancercellsinneboende mekanism. Denna slutsats stöds särskilt av analysen av 198 cellinjer som representerar 22 cancertyper, inklusive SCC, där 12 stabilt heterogena epigenetiska tillstånd (inklusive p-EMT i SCC) detekterades på olika sätt i cellinjemodellerna såväl som deras relaterade primära tumörer (75).
Återigen kunde de heterogena fenotypiska tillstånden inte kopplas till detekterbara genetiska skillnader, och i flera fall har FACS-sorterade celler i ett visst tillstånd visats dynamiskt återutjämnas vid odling, vilket återskapar en stabil jämvikt mellan de heterogena tillstånden som observerats i de ursprungliga cellinjerna.
Vidare belyser teknologier för genomomfattande profilering av olika attribut – bortom DNA-sekvensen och dess mutationsvariation – inflytelserika delar av annoteringen och organisationen av cancercellsgenomet som korrelerar med patientens prognos och, i allt högre grad, med karakteristiska förmågor (76-78). Epigenomisk heterogenitet avslöjas av allt kraftfullare teknologier för profilering av genomomfattande DNA-metylering (79, 80), histonmodifiering (81), kromatintillgänglighet (82) och post-transkriptionell modifiering och translation av RNA (83, 84).
En utmaning med avseende på postulatet som betraktas här kommer att vara att bestämma vilka epigenomiska modifieringar i vissa cancertyper (i) har regulatorisk betydelse och (ii) är representativa för rent icke-mutationell omprogrammering, i motsats till mutationsdriven och därmed genomförklarbar instabilitet.
Epigenetisk reglering av stromacelltyper som befolkar tumörmikromiljön
I allmänhet tros inte de accessoriska cellerna i tumörmikromiljön som funktionellt bidrar till förvärvet av karakteristiska förmågor lida av genetisk instabilitet och mutationell omprogrammering för att förbättra sina tumörfrämjande aktiviteter; snarare dras slutsatsen att dessa celler – cancerassocierade fibroblaster, medfödda immunceller och endotelceller och pericyter i tumörens kärlsystem – omprogrammeras epigenetiskt vid deras rekrytering av lösliga och fysiska faktorer som definierar den solida tumörens mikromiljö (2, 85).
Det förväntas att de multiomiska profileringsteknologier som för närvarande tillämpas på cancerceller kommer att användas alltmer för att studera accessoriska (stromala) celler i tumörer för att klargöra hur normala celler skadas för att funktionellt stödja tumörutveckling och progression. Till exempel tyder en nyligen genomförd studie (86) på att sådan omprogrammering kan involvera modifieringar av epigenomet, förutom det induktiva utbytet av cytokiner, kemokiner och tillväxtfaktorer som förändrar intracellulära signalnätverk i alla dessa celltyper:
När musmodeller med lungmetastaser behandlades med en kombination av en DNA-metyltransferashämmare (5-azacytidin) och en histonmodifieringshämmare (en HDAC), befanns de infiltrerande myeloidcellerna ha övergått från ett omoget (tumörfrämjande) stamfadertillstånd till celler som liknar mogna interstitiala motparter, som liknar deras makrotumörliknande () i obehandlade tumörer, kunde inte stödja de typiska förmågor som krävs för effektiv metastatisk kolonisering (86). Det är tänkbart att multiomisk profilering och farmakologiska störningar kommer att tjäna till att belysa det omprogrammerade epigenetiska tillståndet i sådana myeloidceller såväl som andra karakteristiska accessoriska celltyper som befolkar tumörmikromiljöer.
Sammanfattning
Tillsammans stöder dessa illustrativa ögonblicksbilder tesen att epigenetisk omprogrammering utan mutation kommer att accepteras som en sann möjliggörande egenskap som tjänar till att underlätta förvärvet av karakteristiska förmågor (Fig. 3), skild från genomisk DNA-instabilitet och mutation. I synnerhet kan icke-mutationell epigenetisk omprogrammering förväntas visa sig vara en integrerad del för att möjliggöra den preliminära nya distinkta förmågan hos fenotypisk plasticitet som diskuterats ovan, särskilt som en drivkraft i den dynamiska transkriptomiska heterogeniteten som blir allt mer väldokumenterad i maligna cancerceller TME. Framstegen för encelliga multiomiska profileringsteknologier kommer att belysa respektive bidrag och samspel mellan mutationsdriven och icke-mutationsdriven epigenetisk reglering i utvecklingen av tumörer under malign progression och metastasering.
Polymorfa mikrobiomer
En långtgående gräns inom biomedicin utvecklas genom att belysa mångfalden och variationen i överflödet av mikroorganismer, gemensamt kallade mikrobiotan, som associeras symbiotiskt med kroppens barriärvävnader som exponeras för den yttre miljön - särskilt epidermis och inre slemhinnor i mag-tarmkanalen, såväl som i mag-tarmkanalen och i lungorna och tarmarna.
Det finns en ökad insikt om att de ekosystem som skapas av inhemska bakterier och svampar – mikrobiomen – har djupgående effekter på hälsa och sjukdom (87), en insikt som drivs av förmågan att screena populationerna av mikrobiella arter med hjälp av nästa generations sekvenserings- och bioinformatikteknologier. För cancer blir bevisen alltmer övertygande att polymorf variabilitet i mikrobiomen mellan individer i en population kan ha djupgående effekter på cancerfenotyper (88, 89).
Associationsstudier i mänskliga och experimentella manipulationer i musmodeller av cancer avslöjar vissa mikroorganismer, främst men inte uteslutande bakterier, som kan ha antingen skyddande eller skadliga effekter på cancerutveckling, malign progression och svar på terapi. Detta gäller även den globala komplexiteten och sammansättningen av ett vävnadsmikrobiom som helhet. Medan tarmmikrobiomet var pionjären för denna nya gräns, har flera vävnader och organ associerat mikrobiomer som uppvisar särdrag relaterade till populationsdynamik och mångfalden av mikrobiella arter och underarter.
Denna växande insikt om vikten av polymorfiskt variabla mikrobiomer för hälsa och sjukdom väcker frågan: Är mikrobiomet en distinkt möjliggörande egenskap som i stort sett påverkar, både positivt och negativt, förvärvet av distinkta förmågor för cancer? Jag överväger denna möjlighet nedan och illustrerar bevis för några av de framträdande vävnadsmikrobiomen som är inblandade i canceregenskaper (Fig. 4), med början på den mest framträdande och uppenbarligen mest påverkande mikrobiomen, den i tarmkanalen.
Figur 4
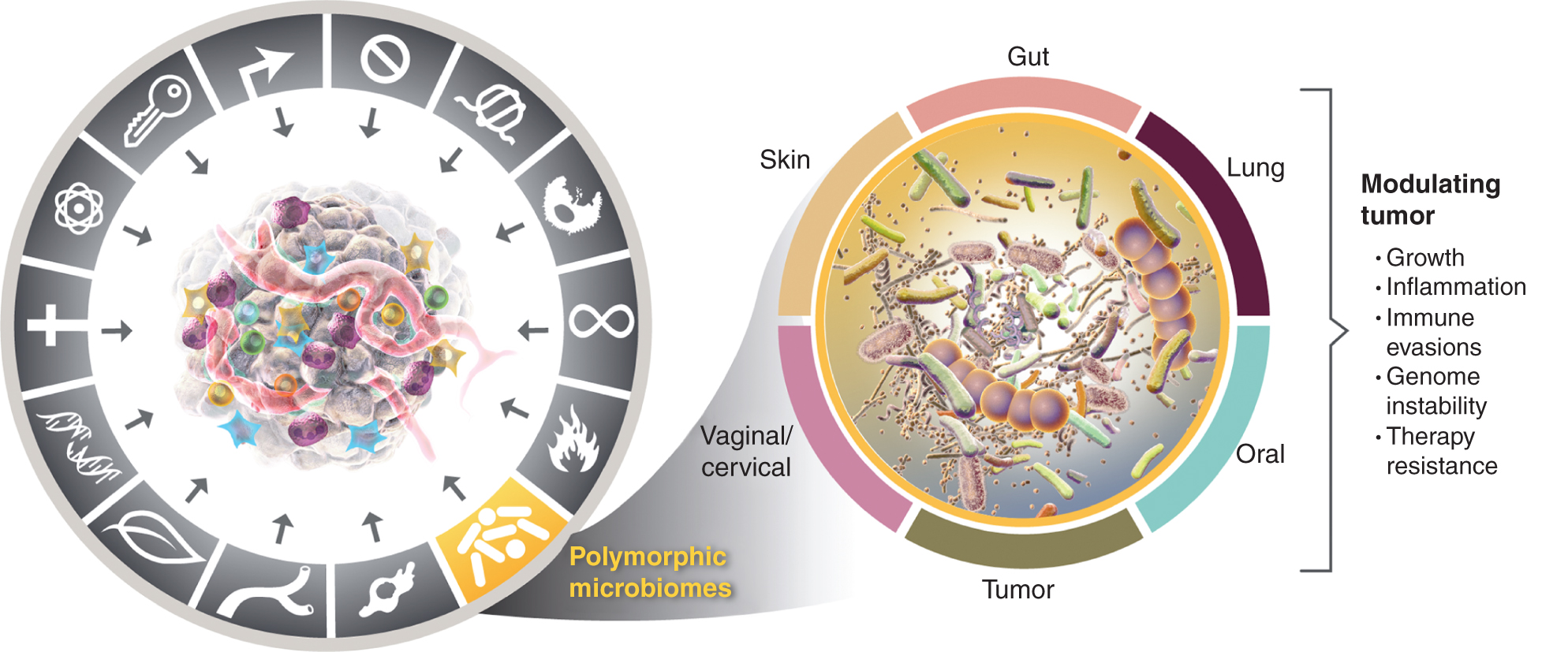
Till vänster, medan de möjliggörande egenskaperna hos tumörfrämjande inflammation och genomisk instabilitet och mutation överlappar varandra, finns det allt större skäl att dra slutsatsen att polymorfa mikrobiomer lokaliserade hos en individ jämfört med en annan i tjocktarmen, i andra slemhinnor och associerade organ, eller i själva tumörerna, kan påverka många av de karakteristiska förmågorna på en mängd olika sätt - antingen genom induktion och inhibering eller kan därför vara inhiberande eller inhiberande. pussel om hur cancer utvecklas, fortskrider och växer svarar på terapi. Det är sant att flera vävnadsmikrobiomer är involverade i att modulera tumörfenotyper. Förutom det brett studerade tarmmikrobiomet är andra karakteristiska vävnadsmikrobiomer såväl som tumörmikrobiomet involverade i att modulera förvärvet - både positivt och negativt - av de karakteristiska förmågor som presenteras i vissa tumörtyper. Varumärkena för cancergrafiken antogs från Hanahan och Weinberg (2).
Flera modulerande effekter av tarmmikrobiomet
Det har länge varit känt att tarmmikrobiomet är grundläggande för tjocktarmens (tjocktarmen) funktion när det gäller att bryta ner och importera näringsämnen till kroppen som en del av metabol homeostas, och att störningar av mikrobiella populationer – dysbios – i tjocktarmen kan orsaka ett spektrum av fysiologiska sjukdomar (87). Detta inkluderar misstanken om att känsligheten, utvecklingen och patogenesen av tjocktarmscancer påverkas av tarmmikrobiomet. Under de senaste åren har övertygande funktionella studier med fekala transplantationer från tjocktarmstumörbärande patienter och möss till mottagarmöss som är predisponerade för utveckling av tjocktarmscancer etablerat en princip: det finns både cancerskyddande och tumörfrämjande mikrobiomer som involverar specifika bakteriearter som kan modulera förekomsten och patogenesen av tjocktarmstumörer (90).
Mekanismerna genom vilka mikrobiota ger dessa modulerande roller klargörs fortfarande, men två generella effekter är alltmer väletablerade för tumörfrämjande mikrobiomer och, i vissa fall, för specifika tumörfrämjande bakteriearter. Den första effekten är mutagenes av tjocktarmsepitelet som ett resultat av produktionen av bakteriella toxiner och andra molekyler som antingen direkt skadar DNA eller stör systemen som upprätthåller genomisk integritet eller på annat sätt stressar celler, vilket indirekt påverkar troheten för DNA-replikation och reparation. Ett typiskt exempel är E. coli, som bär PKS-lokuset, som har visat sig mutagenisera det mänskliga genomet och är involverat i överföringen av mutationer som möjliggör märket (91).
Vidare har bakterier rapporterats binda till ytan av kolonitepitelceller och producera ligandmimetika som stimulerar epitelproliferation, vilket bidrar till den karakteristiska proliferativa signaleringsförmågan i neoplastiska celler (88). En annan mekanism genom vilken specifika typer av bakterier främjar tumörutveckling är butyratproducerande bakterier, vars förekomst ökar hos patienter med kolorektal cancer (92).
Produktionen av metaboliten butyrat har komplexa fysiologiska effekter, inklusive induktion av senescenta epitel- och fibroblastceller. En musmodell av koloncarcinogenes koloniserad med butyratproducerande bakterier utvecklade fler tumörer än möss som saknade sådana bakterier; Sambandet mellan butyratinducerad senescens och ökad kolontumörbildning har visats genom användning av ett senolytiskt läkemedel som dödar åldrande celler, vilket försämrar tumörtillväxt (92).
Vidare har bakteriellt producerat butyrat pleiotropa och paradoxala effekter på differentierade celler jämfört med odifferentierade (stam)celler i tjocktarmsepitelet under tillstånd där tarmbarriären är avbruten (dysbios) och bakterierna är invasiva, vilket påverkar till exempel cellulär energi och metabolism, histonprogress-immunifiering, och (inflammation av histon) som immunsuppressar adaptiva immunsvar (93).
Faktum är att en bred verkan av polymorfa mikrobiomer involverar modulering av det adaptiva och medfödda immunsystemet genom olika vägar, inklusive produktion av "immunomodulerande" faktorer av bakterier som aktiverar skadesensorer på epiteliala eller inhemska immunceller, vilket leder till uttryck av en mångfald repertoar av kemokiner och cytokiner som kan forma immuncellerna och epitoniska cellerna. underliggande stroma och dränerande lymfkörtlar.
Dessutom kan vissa bakterier bryta mot både den skyddande biofilmen och slemmet som täcker tjocktarmsepitelet och störa de täta förbindelserna mellan epitelceller och celler som tillsammans upprätthåller integriteten hos den fysiska barriären som normalt delar upp tarmmikrobiomet. Vid invadering av stroma kan bakterier utlösa både medfödda och adaptiva immunsvar genom att framkalla utsöndring av en repertoar av cytokiner och kemokiner. En manifestation kan vara skapandet av tumörfrämjande eller tumörantagoniserande immunmikromiljöer, som följaktligen skyddar mot eller underlättar tumörbildning och malign progression.
Följaktligen kan moduleringen av de sammanflätade parametrarna för (i) induktionen av (medfödd) tumörbefrämjande inflammation och (ii) flykten från (adaptiv) immunförstöring av karakteristiska mikrobiomer hos individuella patienter vara associerad inte bara med prognos utan också med svar eller resistens mot immunterapier med immunkontrollpunktshämmare och andra terapeutiska manifestationer av tumörer och andra terapeutiska manifestationer. tumörantagoniserande immunmikromiljöer, som följaktligen uppstår Skydda eller underlätta tumörutveckling och malign progression.
Följaktligen kan moduleringen av de sammanflätade parametrarna för (i) induktionen av (medfödd) tumörbefrämjande inflammation och (ii) flykten från (adaptiv) immunförstöring av karakteristiska mikrobiomer hos individuella patienter vara associerad inte bara med prognos utan också med svar eller resistens mot immunterapier med immunkontrollpunktshämmare och andra terapeutiska manifestationer av tumörer och andra terapeutiska manifestationer. tumörantagoniserande immunmikromiljöer, som följaktligen uppstår skyddar eller underlättar tumörutveckling och malign progression).
Följaktligen kan modulering av de sammanflätade parametrarna för (i) induktion av (medfödd) tumörfrämjande inflammation och (ii) flykt från (adaptiv) immunförstöring av distinkta mikrobiomer hos enskilda patienter vara associerad inte bara med prognos utan också med svar eller resistens mot immunterapier med immunkontrollpunktshämmare och andra terapeutiska modaliteter (4–96, 96). Preliminär proof-of-concept kommer från nya studier som visar återställd effekt av immunterapi efter transplantationer av fekal mikrobiota från terapiresponders till patienter med melanom som hade utvecklats under tidigare behandling med immunkontrollpunktsblockad (97, 98).
De molekylära mekanismerna genom vilka distinkta och variabla komponenter i tarmmikrobiomet systemiskt modulerar aktiviteten hos det adaptiva immunsystemet förblir ett ihållande mysterium, antingen genom att förstärka antitumörimmunsvar framkallade av immunkontrollpunktsblockad eller snarare genom att inducera systemisk eller lokal (intratumoral) immunsuppression. En nyligen genomförd studie har kastat ljus: vissa stammar av Enterococcus (och andra bakterier) uttrycker ett peptidoglykanhydrolyas som kallas SagA, som frisätter mukopeptider från bakterieväggen, som sedan kan cirkulera systemiskt och aktivera NOD2-mönsterreceptorn, vilket i sin tur ökar T-cellssvaren och effektiviteten av checkpoint-immunterapi (99).
Andra immunreglerande molekyler som produceras av specifika bakteriella underarter identifieras och funktionellt bedöms, inklusive bakterieproducerat inosin, en hastighetsbegränsande metabolit för T-cellsaktivitet (100). Dessa och andra exempel börjar avgränsa de molekylära mekanismerna genom vilka polymorfa mikrobiomer indirekt och systemiskt modulerar tumörimmunobiologi, utöver immunsvar som följer direkta fysiska interaktioner mellan bakterier och immunsystemet (101, 102).
Bortsett från orsakssambanden till tjocktarmscancer och melanom, är den påvisade förmågan hos tarmmikrobiomet att framkalla uttryck av immunmodulerande kemokiner och cytokiner som kommer in i den systemiska cirkulationen också uppenbarligen kapabel att påverka cancerpatogenes och svar på terapier i andra organ i kroppen (94, 95).
Ett belysande exempel rör utvecklingen av kolangiokarcinom i levern: intestinal dysbios tillåter inträde och transport av bakterier och bakteriella produkter genom portvenen till levern, där TLR4 uttryckt på hepatocyter triggas för att inducera uttryck av kemokinen CXCL1, som rekryterar CXSC-expressande cellceller som tjänar CXCR2-expressiva celler. undertrycka naturliga mördarceller för att undvika immunförstöring (103) och förmodligen förmedla andra distinkta förmågor (85). Som sådan är tarmmikrobiomet tydligt inblandat som en möjliggörande funktion som alternativt kan underlätta eller skydda mot flera cancerformer.
Bortom tarmen: Implicerar distinkta mikrobiomer i andra barriärvävnader
Nästan alla vävnader och organ som är direkt eller indirekt exponerade för den yttre miljön är också förråd av kommensala mikroorganismer (104). I motsats till tarmen, där mikrobiomens symbiotiska roll i ämnesomsättningen är välkänd, framträder fortfarande de normala och patogena rollerna för den inhemska mikrobiotan på dessa olika platser.
Det finns uppenbara organ/vävnadsspecifika skillnader i konstitutionen av de associerade mikrobiomen i homeostas, åldrande och cancer, med både överlappande och distinkta arter och frekvenser till de i tjocktarmen (104, 105). Dessutom ger associationsstudier allt större bevis för att lokala tumörmotverkande/skyddande kontra tumörfrämjande vävnadsmikrobiomer, liknande tarmmikrobiomet, kan modulera känslighet och patogenes för humana cancerformer som uppstår i deras associerade organ (106-109).
Påverkan av den intratumorala mikrobiotan?
Slutligen har patologer länge insett att bakterier kan detekteras i solida tumörer, en observation som nu har underbyggts av sofistikerad profileringsteknik. Till exempel, i en studie av 1 526 tumörer som spänner över sju mänskliga cancertyper (ben, hjärna, bröst, lunga, melanom, äggstockar och bukspottkörtel), kännetecknades varje typ av en distinkt mikrobiom, som till stor del finns i cancerceller och immunceller. Inom varje tumörtyp har variationer i tumörmikrobiomet visats och kommit fram till att de är associerade med klinikopatologiska egenskaper (110).
Mikrobiota har på liknande sätt upptäckts i de novo genetiskt modifierade musmodeller av lung- och bukspottkörtelcancer, och deras frånvaro i bakteriefria möss och/eller deras upphävande med antibiotika kan visa sig försämra tumörbildningen, vilket funktionellt implicerar tumörmikrobiomet som en prekursor till tumörfrämjande inflammation och malign progression11211, elakartad progression11211.
Associationsstudier i humant pankreatiskt duktalt adenokarcinom och funktionella analyser via fekal transplantation i tumörbärande möss har visat att variationer i tumörmikrobiomet – och det associerade tarmmikrobiomet – modulerar immunsystemets fenotyper och överlevnad (113). En viktig utmaning för framtiden kommer att vara att utvidga dessa implikationer till andra tumörtyper och att distrahera de potentiellt separerbara bidragen av konstitution och variation i tumörmikrobiomet från tarmmikrobiomets (och den lokala ursprungsvävnaden), kanske genom att identifiera specifika mikrobiella arter som är funktionellt inflytelserika på ett eller annat ställe.
Sammanfattning
Spännande frågor för framtiden inkluderar huruvida mikrobiota som finns i olika vävnader eller befolkar begynnande neoplasmer har förmågan att bidra till eller störa förvärvet av andra distinkta förmågor utöver immunmodulering och genomisk mutation, och därigenom påverka tumörutveckling och progression. Det finns bevis för att vissa bakteriearter direkt kan stimulera kännetecknet för proliferativ signalering, till exempel i tjocktarmsepitelet (88), och kan modulera tillväxthämning genom att förändra tumörsuppressoraktiviteten i olika delar av tarmen (114), samtidigt som de har direkta effekter på andra karakteristiska förmågor, såsom att undvika celldöd, trigga, utlösa, stimulera angiogenes och kvarstå. generaliserbarheten av dessa observationer till flera former av mänsklig cancer.
Oavsett vilket finns det alltmer övertygande argument för att polymorf variation i mikrobiomen i tarmen och andra organ representerar en distinkt aktiverande egenskap för förvärvet av distinkta färdigheter (Fig. 4), även om det överlappar med och kompletterar genomets instabilitet och mutation, och tumörfrämjande inflammation.
Åldrande celler
Cellulär senescens är en typiskt irreversibel form av proliferativ stopp som sannolikt utvecklats som en skyddande mekanism för att upprätthålla vävnadshomeostas, skenbart som en komplementär mekanism till programmerad celldöd som tjänar till att inaktivera och, i sinom tid, avlägsna sjuka, dysfunktionella eller på annat sätt onödiga celler. Förutom att stänga av celldelningscykeln, producerar senescensprogrammet förändringar i cellmorfologi och metabolism och, allra djupast, aktiveringen av en senescensassocierad sekretorisk fenotyp (SASP), som involverar frisättning av en uppsjö av bioaktiva proteiner, inklusive kemokiner.
Cytokiner och proteaser, vilkas identitet beror på vilken cell och vävnadstyp från vilken en åldrande cell uppstår ( 115–117). Åldrande kan induceras i celler av en mängd olika tillstånd, inklusive mikromiljöpåfrestningar såsom näringssvält och DNA-skador, såväl som skador på organeller och cellulär infrastruktur och obalanser i cellulära signalnätverk (115, 117), som alla har inträffat i samband med den observerade ökningen av frekvensen av senescenta celler vid olika organeller (1) 119).
Cellulär senescens har länge ansetts vara en skyddande mekanism mot neoplasi, vilket gör att cancerceller genomgår åldrande (120). De flesta av de ovan nämnda initiativtagarna till senescensprogrammet är förknippade med malignitet, särskilt DNA-skador till följd av avvikande hyperproliferation, så kallad onkogeninducerad senescens på grund av hyperaktiverad signalering och terapiinducerad åldrande till följd av cellulära och genomiska skador orsakade av kemoterapi och strålbehandling.
Det finns faktiskt väletablerade exempel på de skyddande fördelarna med senescens för att begränsa malign progression (118, 119). Tvärtom visar dock en växande mängd bevis precis motsatsen: i vissa sammanhang stimulerar åldrande celler differentiellt tumörutveckling och malign progression (119, 121).
I en insiktsfull fallstudie, ablerades åldrande celler i åldrande möss farmakologiskt, vilket specifikt utarmade åldrande celler som karakteristiskt uttrycker cellcykelhämmaren p16 – INK4a: förutom att fördröja flera åldersrelaterade symtom, resulterade detta i utarmning av åldrande celler i åldrande möss med minskad incidens-2 cancerdöd (1).
Den huvudsakliga mekanismen genom vilken åldrande celler främjar tumörfenotyper tros vara SASP, som har visat sig kunna förmedla signalmolekyler (och proteaser som aktiverar och/eller deaktiverar) på ett parakrint sätt för att förmedla typiska förmågor. I olika experimentella system har åldrande cancerceller således visat sig bidra på olika sätt till proliferativ signalering, undvika apoptos, inducera angiogenes, stimulera invasion och metastasering och undertrycka tumörimmunitet (116, 118, 120, 121).
Ännu en aspekt av effekterna av åldrande cancerceller på cancerfenotyper involverar övergående, reversibla åldrande celltillstånd, varvid åldrande cancerceller kan undkomma sitt SASP-uttryckande, icke-proliferativa tillstånd och återuppta cellproliferation och manifestation av de associerade egenskaperna hos en fullt livsduglig onkogencell (44).
Sådan övergående åldrande är bäst dokumenterad i fall av terapiresistens (44), som representerar en form av stillastående som undviker terapeutisk inriktning av prolifererande cancerceller, men kan visa sig mer allmänt effektiv i andra stadier av tumörutveckling, malign progression och metastasering.
De senescenta cellernas känneteckenfrämjande förmågor är dessutom inte begränsade till senescenta cancerceller. Cancerassocierade fibroblaster (CAF) har visat sig åldras i tumörer, vilket ger upphov till åldrande CAF som har visat sig vara tumörfrämjande genom att ge karaktäristiska förmågor till cancerceller i TME (115, 116, 121).
Vidare är åldrande fibroblaster i normala vävnader, delvis bildade av naturligt åldrande eller miljöförolämpningar, på liknande sätt involverade i ombyggnad av vävnadsmikromiljöer via deras SASP för att ge parakrint stöd för lokal invasion (så kallade "fälteffekter") och fjärrmetastaser (116) av neoplasmer som utvecklas i närheten.
Vidare har åldrande fibroblaster i åldrande hud visat sig rekrytera – via sina SASP – medfödda immunceller som både är immunsuppressiva mot adaptiva antitumörimmunsvar förankrade av CD8 T-celler och stimulerar tillväxt av hudtumörer (123), den senare effekten återspeglar möjligen parakrina bidrag från sådana medfödda immunceller och neutrofila egenskaper till andra egenskaper, neutrofila celler och neutrofila celler. reflekterar.
Även om det är mindre väletablerat verkar det troligt att andra rikliga stromaceller som befolkar specifika tumörmikromiljöer kommer att genomgå åldrande, och därigenom modulera canceregenskaper och resulterande tumörfenotyper. Till exempel kan terapiinducerade åldrande tumörendotelceller öka proliferation, invasion och metastasering i bröstcancermodeller (124, 125).
Visst, sådana bevis motiverar utredning i andra tumörtyper för att utvärdera allmän åldrande av fibroblaster, endotelceller och andra stromaceller som en drivkraft i tumörutveckling. För närvarande är också oklara regleringsmekanismer och funktionella bestämningsfaktorer genom vilka en viss åldrande celltyp i en viss TME framkallar en tumörbefrämjande kontra en tumörantagoniserande SASP, som uppenbarligen kan induceras alternativt i samma åldrande celltyp, kanske av olika initiatorer när nedsänkt i karakteristiska fysiologiska och neoplastiska mikromiljöer.
Sammanfattning
Konceptet att tumörer består av genetiskt transformerade cancerceller som interagerar med och drar nytta av rekryterade och epigenetiskt/fenotypiskt korrupta accessoriska (stromala) celler har fastställts som avgörande för patogenesen av cancer. De överväganden som diskuterats ovan och beskrivs i de recensioner och rapporter som citeras här (och på andra ställen) argumenterar övertygande för att åldrande celler (oavsett cellulärt ursprung) bör övervägas för inkludering i listan över funktionellt signifikanta celler i tumörens mikromiljö (Fig. 5). Därför bör åldrande celler övervägas i sökandet efter fördjupad kunskap om cancermekanismer. Vidare motiverar erkännandet av deras betydelse det sekundära målet att terapeutiskt rikta in sig på tumörfrämjande åldrande celler av alla konstitutioner, vare sig det är genom farmakologisk eller immunologisk ablation eller genom att omprogrammera SASP till tumörantagoniserande varianter (115, 121, 126).
Bild 5

Heterogena cancercellsubtyper och stromacelltyper och subtyper är funktionellt integrerade i tumörernas manifestationer som illegala organ. Ökande bevis tyder på att åldrande cellderivat av många av dessa cellulära komponenter i TME och deras variabla SASPs är involverade i moduleringen av känneteckenförmågor och resulterande tumörfenotyper. Varumärkena för cancergrafiken antogs från Hanahan och Weinberg (2).
Avslutande kommentarer
Även om de åtta kännetecknen för cancer och deras två stödjande egenskaper har visat sig ha bestående heuristiskt värde i konceptualiseringen av cancer, tyder de överväganden som presenterats ovan på att det kan finnas nya aspekter av en viss allmänhet och därför betydelse för en mer fullständig förståelse av sjukdomens komplexitet, mekanismer och manifestationer. Genom att tillämpa måttet för urskiljbart, om inte fullständigt, oberoende från de 10 kärnattributen, kan man argumentera för att dessa fyra parametrar - vid ytterligare validering och generalisering utöver de presenterade fallstudierna - mycket väl kan integreras i cancerschemats kännetecken (Fig. 6).
Därför kan cellplasticitet läggas till listan över framträdande funktioner. Medan den åttonde kärnan och denna nyskapande förmåga var och en begreppsmässigt kan särskiljas genom sin definition som kännetecken, är aspekter av deras reglering åtminstone delvis kopplade i vissa och kanske många cancerformer. Till exempel moduleras flera kännetecken koordinerat av kanoniska onkogena drivkrafter i vissa tumörtyper, inklusive
- (I) KRAS ( https://cancer.sanger.ac.uk/cosmic/census-page/KRAS ),
- (II) MYC ( https://cancer.sanger.ac.uk/cosmic/census-page/MYC ),
- (III) NOTCH ( https://cancer.sanger.ac.uk/cosmic/census-page/NOTCH1 ; Ref. 127) und
- (IV) TP53 ( https://cancer.sanger.ac.uk/cosmic/census-page/TP53 )
Bild 6
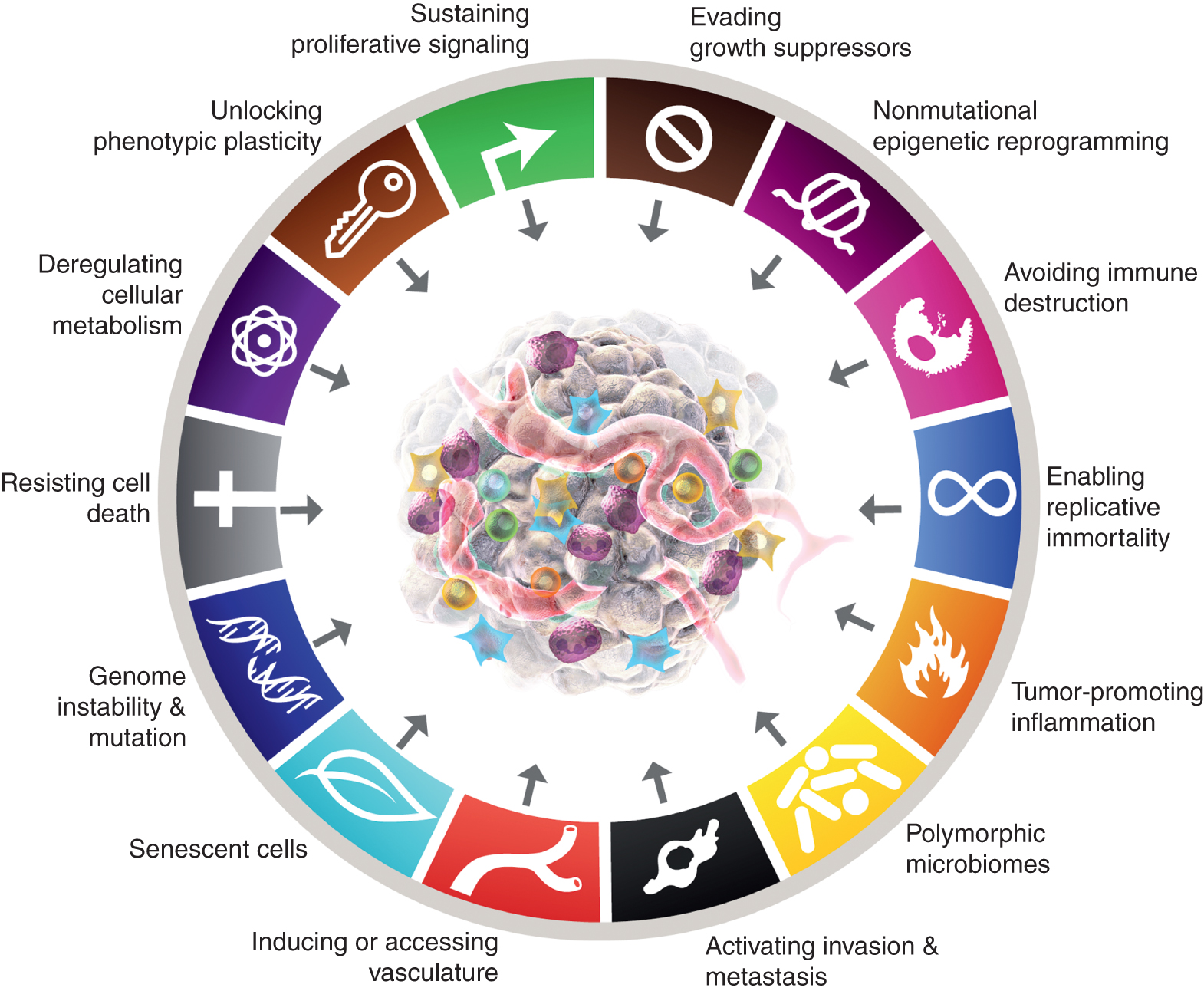
De kanoniska och förväntade nya tilläggen till "Kräftans kännetecken" visas. Detta dokument lyfter möjligheten, i syfte att stimulera debatt, diskussion och experimentell utarbetning, att några eller alla av de fyra nya parametrarna kommer att erkännas som generiska för flera former av cancer hos människor och därför lämpliga för integration i kärnkonceptualiseringen av cancers kännetecken. Varumärkena för cancergrafiken antogs från Hanahan och Weinberg (2).
Förutom att lägga till cellulär plasticitet till förteckningen, kan icke-mutationell epigenetisk omprogrammering och polymorfa variationer integreras i organ/vävnadsmikrobiomer som mekanistiska bestämningsfaktorer – möjliggörande egenskaper – genom vilka distinkta förmågor förvärvas, tillsammans med tumörfrämjande inflammation (som i sig är delvis sammankopplad med mikrobiomet), utöver de ovan nämnda mutationerna och andra faktorer som förmår att manifestera.
Slutligen kan åldrande celler av olika ursprung - inklusive cancerceller och olika stromaceller - som funktionellt bidrar till utvecklingen och maligna progression av cancer, om än på markant olika sätt från deras icke-åldrande bröder, inkluderas som generiska komponenter i TME. Sammanfattningsvis är det tänkt att utplaceringen av dessa preliminära "experimentella ballonger" kommer att stimulera debatt, diskussion och ytterligare experimentell undersökning i cancerforskningssamhället om de definierande begreppsparametrarna för cancerbiologi, genetik och patogenes.
Referenser
- Hanahan D , Weinberg RA . The hallmarks of cancer. Cell 2000;100:57–70.
- Hanahan D , Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Yuan S , Norgard RJ , Stanger BZ . Cellular plasticity in cancer. Cancer Discov 2019;9:837–51.
- Barker N , Ridgway RA , van Es JH , van de Wetering M , Begthel H , van den Born M et al . Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608–11.
- Perekatt AO , Shah PP , Cheung S , Jariwala N , Wu A , Gandhi V et al . SMAD4 suppresses WNT-driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res 2018;78:4878–90.
- Shih IM , Wang TL , Traverso G , Romans K , Hamilton SR , Ben-Sasson S et al . Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A 2001;98:2640–5.
- Ordóñez-Morán P , Dafflon C , Imajo M , Nishida E , Huelsken J . HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 2015;28:815–29.
- Tan SH , Barker N . Stemming colorectal cancer growth and metastasis: HOXA5 forces cancer stem cells to differentiate. Cancer Cell 2015;28:683–5.
- Köhler C , Nittner D , Rambow F , Radaelli E , Stanchi F , Vandamme N et al . Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 2017;21:679–93.
- Shah M , Bhoumik A , Goel V , Dewing A , Breitwieser W , Kluger H et al . A role for ATF2 in regulating MITF and melanoma development. PLoS Genet 2010;6:e1001258.
- Claps G , Cheli Y , Zhang T , Scortegagna M , Lau E , Kim H et al . A transcriptionally inactive ATF2 variant drives melanomagenesis. Cell Rep 2016;15:1884–92.
- Saghafinia S , Homicsko K , Di Domenico A , Wullschleger S , Perren A , Marinoni I et al . Cancer cells retrace a stepwise differentiation program during malignant progression. Cancer Discov 2021;11:2638–57.
- Yu X-X , Qiu W-L , Yang L , Zhang Y , He M-Y , Li L-C et al . Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J 2019;38:e100164.
- de Thé H . Differentiation therapy revisited. Nat Rev Cancer 2018;18:117–27.
- He LZ , Merghoub T , Pandolfi PP . In vivo analysis of the molecular pathogenesis of acute promyelocytic leukemia in the mouse and its therapeutic implications. Oncogene 1999;18:5278–92.
- Warrell RP , de Thé H , Wang ZY , Degos L . Acute promyelocytic leukemia. N Engl J Med 1993;329:177–89.
- Bots M , Verbrugge I , Martin BP , Salmon JM , Ghisi M , Baker A et al . Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014;123:1341–52.
- Ferrara FF , Fazi F , Bianchini A , Padula F , Gelmetti V , Minucci S et al . Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res 2001;61:2–7.
- Kaufman CK , Mosimann C , Fan ZP , Yang S , Thomas AJ , Ablain J et al . A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016;351:aad2197.
- Morris JP , Yashinskie JJ , Koche R , Chandwani R , Tian S , Chen C-C et al . α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019;573:595–9.
- Saha SK , Parachoniak CA , Ghanta KS , Fitamant J , Ross KN , Najem MS et al . Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110–4.
- Dang L , Su S-SM . Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem 2017;86:305–31.
- Waitkus MS , Diplas BH , Yan H . Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018;34:186–95.
- Phan TG , Croucher PI . The dormant cancer cell life cycle. Nat Rev Cancer 2020;20:398–411.
- Jiang M , Azevedo-Pouly AC , Deering TG , Hoang CQ , DiRenzo D , Hess DA et al . MIST1 and PTF1 collaborate in feed-forward regulatory loops that maintain the pancreatic acinar phenotype in adult mice. Mol Cell Biol 2016;36:2945–55.
- Krah NM , Narayanan SM , Yugawa DE , Straley JA , Wright CVE , MacDonald RJ et al . Prevention and reversion of pancreatic tumorigenesis through a differentiation-based mechanism. Dev Cell 2019;50:744–54.
- Krah NM , De La O J-P , Swift GH , Hoang CQ , Willet SG , Chen Pan F et al . The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015;4:e07125.
- Shi G , DiRenzo D , Qu C , Barney D , Miley D , Konieczny SF . Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 2013;32:1950–8.
- Kopp JL , von Figura G , Mayes E , Liu F-F , Dubois CL , Morris JP et al . Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50.
- Julian LM , McDonald AC , Stanford WL . Direct reprogramming with SOX factors: masters of cell fate. Curr Opin Genet Dev 2017;46:24–36.
- Grimm D , Bauer J , Wise P , Krüger M , Simonsen U , Wehland M et al . The role of SOX family members in solid tumours and metastasis. Semin Cancer Biol 2020;67:122–53.
- Mu P , Zhang Z , Benelli M , Karthaus WR , Hoover E , Chen C-C et al . SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84–8.
- Von Hoff DD , LoRusso PM , Rudin CM , Reddy JC , Yauch RL , Tibes R et al . Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72.
- Biehs B , Dijkgraaf GJP , Piskol R , Alicke B , Boumahdi S , Peale F et al . A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018;562:429–33.
- Boumahdi S , de Sauvage FJ . The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19:39–56.
- Groves SM , Ireland A , Liu Q , Simmons AJ , Lau K , Iams WT et al . Cancer Hallmarks Define a Continuum of Plastic Cell States between Small Cell Lung Cancer Archetypes [Internet]. Systems Biology; 2021 Jan. Available from: http://biorxiv.org/lookup/doi/10.1101/2021.01.22.427865.
- LaFave LM , Kartha VK , Ma S , Meli K , Del Priore I , Lareau C et al . Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 2020;38:212–28.
- Marjanovic ND , Hofree M , Chan JE , Canner D , Wu K , Trakala M et al . Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 2020;38:229–46.
- Drapkin BJ , Minna JD . Studying lineage plasticity one cell at a time. Cancer Cell 2020;38:150–2.
- Inoue Y , Nikolic A , Farnsworth D , Liu A , Ladanyi M , Somwar R et al . Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer [Internet]. Cancer Biology; 2020 Nov. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.11.12.368522.
- Dravis C , Chung C-Y , Lytle NK , Herrera-Valdez J , Luna G , Trejo CL et al . Epigenetic and transcriptomic profiling of mammary gland development and tumor models disclose regulators of cell state plasticity. Cancer Cell 2018;34:466–82.
- Malta TM , Sokolov A , Gentles AJ , Burzykowski T , Poisson L , Weinstein JN et al . Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–54.
- Miao Z-F , Lewis MA , Cho CJ , Adkins-Threats M , Park D , Brown JW et al . A dedicated evolutionarily conserved molecular network licenses differentiated cells to return to the cell cycle. Dev Cell 2020;55:178–94.
- De Blander H , Morel A-P , Senaratne AP , Ouzounova M , Puisieux A . Cellular plasticity: a route to senescence exit and tumorigenesis. Cancers 2021;13:4561.
- Merrell AJ , Stanger BZ . Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 2016;17:413–25.
- Baylin SB , Jones PA . Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 2016;8:a019505.
- Flavahan WA , Gaskell E , Bernstein BE . Epigenetic plasticity and the hallmarks of cancer. Science 2017;357:eaal2380.
- Jones PA , Issa J-PJ , Baylin S . Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630–41.
- Huang S . Tumor progression: Chance and necessity in Darwinian and Lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol 2012;110:69–86.
- Darwiche N . Epigenetic mechanisms and the hallmarks of cancer: an intimate affair. Am J Cancer Res 2020;10:1954–78.
- Feng Y , Liu X , Pauklin S . 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell 2021;12:440–54.
- Nam AS , Chaligne R , Landau DA . Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet 2021;22:3–18.
- Bitman-Lotan E , Orian A . Nuclear organization and regulation of the differentiated state. Cell Mol Life Sci CMLS 2021;78:3141–58.
- Goldberg AD , Allis CD , Bernstein E . Epigenetics: a landscape takes shape. Cell 2007;128:635–8.
- Zeng Y , Chen T . DNA methylation reprogramming during mammalian development. Genes 2019;10:257.
- Hegde AN , Smith SG . Recent developments in transcriptional and translational regulation underlying long-term synaptic plasticity and memory. Learn Mem 2019;26:307–17.
- Kim S , Kaang B-K . Epigenetic regulation and chromatin remodeling in learning and memory. Exp Mol Med 2017;49:e281.
- Thienpont B , Van Dyck L , Lambrechts D . Tumors smother their epigenome. Mol Cell Oncol 2016;3:e1240549.
- Gameiro PA , Struhl K . Nutrient deprivation elicits a transcriptional and translational inflammatory response coupled to decreased protein synthesis. Cell Rep 2018;24:1415–24.
- Lin GL , Monje M . Understanding the deadly silence of posterior fossa A ependymoma. Mol Cell 2020;78:999–1001.
- Michealraj KA , Kumar SA , Kim LJY , Cavalli FMG , Przelicki D , Wojcik JB et al . Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell 2020;181:1329–45.
- Bakir B , Chiarella AM , Pitarresi JR , Rustgi AK . EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol 2020;30:764–76.
- Gupta PB , Pastushenko I , Skibinski A , Blanpain C , Kuperwasser C . Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 2019;24:65–78.
- Lambert AW , Weinberg RA . Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer 2021;21:325–38.
- Lindner P , Paul S , Eckstein M , Hampel C , Muenzner JK , Erlenbach-Wuensch K et al . EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis 2020;11:147.
- Javaid S , Zhang J , Anderssen E , Black JC , Wittner BS , Tajima K et al . Dynamic chromatin modification sustains epithelial-mesenchymal transition following inducible expression of Snail-1. Cell Rep 2013;5:1679–89.
- Serrano-Gomez SJ , Maziveyi M , Alahari SK . Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 2016;15:18.
- Skrypek N , Goossens S , De Smedt E , Vandamme N , Berx G . Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet TIG 2017;33:943–59.
- Li L , Hanahan D . Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 2013;153:86–100.
- Li L , Zeng Q , Bhutkar A , Galván JA , Karamitopoulou E , Noordermeer D et al . GKAP acts as a genetic modulator of NMDAR signaling to govern invasive tumor growth. Cancer Cell 2018;33:736–51.
- Mohammadi H , Sahai E . Mechanisms and impact of altered tumour mechanics. Nat Cell Biol 2018;20:766–74.
- Odenthal J , Takes R , Friedl P . Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis 2016;37:1117–28.
- Torres CM , Biran A , Burney MJ , Patel H , Henser-Brownhill T , Cohen A-HS et al . The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 2016;353:aaf1644.
- Puram SV , Tirosh I , Parikh AS , Patel AP , Yizhak K , Gillespie S et al . Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171:1611–24.
- Kinker GS , Greenwald AC , Tal R , Orlova Z , Cuoco MS , McFarland JM et al . Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 2020;52:1208–18.
- Murtha M , Esteller M . Extraordinary cancer epigenomics: thinking outside the classical coding and promoter box. Trends Cancer 2016;2:572–84.
- Nebbioso A , Tambaro FP , Dell’Aversana C , Altucci L . Cancer epigenetics: moving forward. PLoS Genet 2018;14:e1007362.
- Tavernari D , Battistello E , Dheilly E , Petruzzella AS , Mina M , Sordet-Dessimoz J et al . Non-genetic evolution drives lung adenocarcinoma spatial heterogeneity and progression. Cancer Discov 2021;11:1490–507.
- Heyn H , Vidal E , Ferreira HJ , Vizoso M , Sayols S , Gomez A et al . Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 2016;17:11.
- Saghafinia S , Mina M , Riggi N , Hanahan D , Ciriello G . Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 2018;25:1066–80.
- Audia JE , Campbell RM . Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8:a019521.
- Corces MR , Granja JM , Shams S , Louie BH , Seoane JA , Zhou W et al . The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898.
- Esteve-Puig R , Bueno-Costa A , Esteller M . Writers, readers and erasers of RNA modifications in cancer. Cancer Lett 2020;474:127–37.
- Janin M , Coll-SanMartin L , Esteller M . Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer 2020;19:70.
- Hanahan D , Coussens LM . Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–22.
- Lu Z , Zou J , Li S , Topper MJ , Tao Y , Zhang H et al . Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020;579:284–90.
- Thomas S , Izard J , Walsh E , Batich K , Chongsathidkiet P , Clarke G et al . The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res 2017;77:1783–812.
- Dzutsev A , Badger JH , Perez-Chanona E , Roy S , Salcedo R , Smith CK et al . Microbes and cancer. Annu Rev Immunol 2017;35:199–228.
- Helmink BA , Khan MAW , Hermann A , Gopalakrishnan V , Wargo JA . The microbiome, cancer, and cancer therapy. Nat Med 2019;25:377–88.
- Sears CL , Garrett WS . Microbes, microbiota, and colon cancer. Cell Host Microbe 2014;15:317–28.
- Pleguezuelos-Manzano C , Puschhof J , Rosendahl Huber A , van Hoeck A , Wood HM , Nomburg J et al . Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020;580:269–73.
- Okumura S , Konishi Y , Narukawa M , Sugiura Y , Yoshimoto S , Arai Y et al . Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat Commun 2021;12:5674.
- Salvi PS , Cowles RA . Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in homeostasis and disease. Cells 2021;10:1775.
- Fessler J , Matson V , Gajewski TF . Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 2019;7:108.
- Gopalakrishnan V , Helmink BA , Spencer CN , Reuben A , Wargo JA . The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 2018;33:570–80.
- Zitvogel L , Ma Y , Raoult D , Kroemer G , Gajewski TF . The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–70.
- Baruch EN , Youngster I , Ben-Betzalel G , Ortenberg R , Lahat A , Katz L et al . Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021;371:602–9.
- Davar D , Dzutsev AK , McCulloch JA , Rodrigues RR , Chauvin J-M , Morrison RM et al . Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021;371:595–602.
- Griffin ME , Espinosa J , Becker JL , Luo J-D , Carroll TS , Jha JK et al . Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 2021;373:1040–6.
- Mager LF , Burkhard R , Pett N , Cooke NCA , Brown K , Ramay H et al . Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369:1481–9.
- Ansaldo E , Belkaid Y . How microbiota improve immunotherapy. Science 2021;373:966–7.
- Ansaldo E , Farley TK , Belkaid Y . Control of immunity by the microbiota. Annu Rev Immunol 2021;39:449–79.
- Zhang Q , Ma C , Duan Y , Heinrich B , Rosato U , Diggs LP et al . Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov 2021;11:1248–67.
- Ding T , Schloss PD . Dynamics and associations of microbial community types across the human body. Nature 2014;509:357–60.
- Byrd AL , Liu M , Fujimura KE , Lyalina S , Nagarkar DR , Charbit B et al . Gut microbiome stability and dynamics in healthy donors and patients with non-gastrointestinal cancers. J Exp Med 2021;218:e20200606.
- Healy CM , Moran GP . The microbiome and oral cancer: more questions than answers. Oral Oncol 2019;89:30-3.
- Swaney MH , Kalan LR . Living in your skin: microbes, molecules and mechanisms. Infect Immun 2021;89:e00695–20.
- Willis JR , Gabaldón T . The human oral microbiome in health and disease: from sequences to ecosystems. Microorganisms 2020;8:308.
- Xu J , Peng J-J , Yang W , Fu K , Zhang Y . Vaginal microbiomes and ovarian cancer: a review. Am J Cancer Res 2020;10:743–56.
- Nejman D , Livyatan I , Fuks G , Gavert N , Zwang Y , Geller LT et al . The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80.
- Jin C , Lagoudas GK , Zhao C , Bullman S , Bhutkar A , Hu B et al . Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019;176:998–1013.
- Pushalkar S , Hundeyin M , Daley D , Zambirinis CP , Kurz E , Mishra A et al . The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 2018;8:403–16.
- McAllister F , Khan MAW , Helmink B , Wargo JA . The tumor microbiome in pancreatic cancer: bacteria and beyond. Cancer Cell 2019;36:577–9.
- Kadosh E , Snir-Alkalay I , Venkatachalam A , May S , Lasry A , Elyada E et al . The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020;586:133–8.
- Birch J , Gil J . Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34:1565–76.
- Faget DV , Ren Q , Stewart SA . Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 2019;19:439–53.
- Gorgoulis V , Adams PD , Alimonti A , Bennett DC , Bischof O , Bishop C et al . Cellular senescence: defining a path forward. Cell 2019;179:813–27.
- He S , Sharpless NE . Senescence in health and disease. Cell 2017;169:1000–11.
- Kowald A , Passos JF , Kirkwood TBL . On the evolution of cellular senescence. Aging Cell 2020;19:e13270.
- Lee S , Schmitt CA . The dynamic nature of senescence in cancer. Nat Cell Biol 2019;21:94–101.
- Wang B , Kohli J , Demaria M . Senescent cells in cancer therapy: friends or foes? Trends Cancer 2020;6:838–57.
- Baker DJ , Childs BG , Durik M , Wijers ME , Sieben CJ , Zhong J et al . Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–9.
- Ruhland MK , Loza AJ , Capietto A-H , Luo X , Knolhoff BL , Flanagan KC et al . Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 2016;7:11762.
- Hwang HJ , Lee Y-R , Kang D , Lee HC , Seo HR , Ryu J-K et al . Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett 2020;490:100–10.
- Wang D , Xiao F , Feng Z , Li M , Kong L , Huang L et al . Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res 2020;22:103.
- Amor C , Feucht J , Leibold J , Ho Y-J , Zhu C , Alonso-Curbelo D et al . Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020;583:127–32.
- Aster JC , Pear WS , Blacklow SC . The varied roles of notch in cancer. Annu Rev Pathol 2017;12:245–75.
- Kuczynski EA , Vermeulen PB , Pezzella F , Kerbel RS , Reynolds AR . Vessel co-option in cancer. Nat Rev Clin Oncol 2019;16:469–93.
Google ScholarCrossref