 Suche
Suche
 Mein Konto
Mein Konto
Sindrome di DiGeorge (sindrome da delezione 22q11.2)
Sindrome di DiGeorge (sindrome da delezione 22q11.2)
panoramica
La sindrome di DiGeorge, meglio conosciuta con un termine più ampio – sindrome da delezione 22q11.2 – è un disturbo causato dalla mancanza di una piccola porzione del cromosoma 22. Questa eliminazione provoca uno scarso sviluppo di diversi sistemi corporei.
Il termine sindrome da delezione 22q11.2 comprende termini che un tempo erano considerati condizioni separate, tra cui la sindrome di DiGeorge, la sindrome velocardiofacciale e altre condizioni che condividono la stessa causa genetica, sebbene le caratteristiche possano variare leggermente.
I problemi medici comunemente associati alla sindrome da delezione 22q11.2 comprendono difetti cardiaci, scarsa funzionalità del sistema immunitario, palatoschisi, complicazioni legate a bassi livelli di calcio nel sangue e ritardo dello sviluppo con problemi comportamentali ed emotivi.
Il numero e la gravità dei sintomi associati alla sindrome da delezione 22q11.2 variano. Tuttavia, quasi tutti coloro che soffrono di questa sindrome necessitano di cure da parte di specialisti in vari campi.
Sintomi
Segni e sintomi della sindrome di DiGeorge (sindrome da delezione 22q11.2) possono variare in tipo e gravità a seconda dei sistemi corporei interessati e della gravità dei difetti. Alcuni segni e sintomi possono essere visibili alla nascita, ma altri potrebbero non comparire fino a tarda infanzia o nella prima infanzia.
Segni e sintomi possono includere una combinazione dei seguenti elementi:
- Herzgeräusch und bläuliche Haut aufgrund schlechter Zirkulation von sauerstoffreichem Blut (Zyanose) als Folge eines Herzfehlers
- Häufige Infektionen
- Bestimmte Gesichtszüge, wie ein unterentwickeltes Kinn, tief angesetzte Ohren, weit auseinanderstehende Augen oder eine schmale Furche in der Oberlippe
- Eine Lücke im Gaumen (Gaumenspalte) oder andere Probleme mit dem Gaumen
- Verzögertes Wachstum
- Schwierigkeiten bei der Nahrungsaufnahme, Gewichtszunahme oder Magen-Darm-Probleme
- Atembeschwerden
- Schlechter Muskeltonus
- Verzögerte Entwicklung, wie z. B. Verzögerungen beim Umdrehen, Aufsetzen oder anderen kindlichen Meilensteinen
- Verzögerte Sprachentwicklung oder nasal klingende Sprache
- Lernverzögerungen oder Behinderungen
- Verhaltensprobleme
Quando andare dal medico?
Altre malattie possono causare segni e sintomi simili alla sindrome da delezione 22q11.2. Pertanto, è importante ottenere una diagnosi accurata e rapida se il bambino mostra uno qualsiasi dei segni o sintomi sopra elencati.
I medici possono sospettare la sindrome da delezione 22q11.2:
- Bei der Geburt. Wenn bestimmte Zustände – ein schwerer Herzfehler, eine Gaumenspalte oder eine Kombination anderer Faktoren, die für das 22q11.2-Deletionssyndrom typisch sind – bereits bei der Geburt offensichtlich sind, werden diagnostische Tests wahrscheinlich beginnen, bevor Ihr Kind das Krankenhaus verlässt.
- Bei wohlauf-Baby-Besuchen. Ihr Hausarzt oder Kinderarzt kann die Störung aufgrund einer Kombination von Krankheiten oder Störungen vermuten, die im Laufe der Zeit auftreten. Andere Probleme können Ihrem Arzt während regelmäßig geplanter Arztbesuche oder jährlicher Vorsorgeuntersuchungen für Ihr Kind auffallen.
Cause
Ogni persona ha due copie del cromosoma 22, una ereditata da ciascun genitore. Quando una persona è affetta dalla sindrome di DiGeorge (sindrome da delezione 22q11.2), in una copia del cromosoma 22 manca un segmento contenente circa 30-40 geni. Molti di questi geni non sono stati chiaramente identificati e non sono ben compresi. La regione deleta del cromosoma 22 è nota come 22q11.2.
La delezione dei geni sul cromosoma 22 di solito si verifica come un evento casuale nello sperma del padre o nell'ovulo della madre, oppure può verificarsi precocemente durante lo sviluppo fetale. In rari casi, la delezione è una malattia ereditaria che viene trasmessa da un genitore a un figlio che presenta anche delezioni nel cromosoma 22 ma può o meno presentare sintomi.
Complicazioni
Difetto del setto ventricolare
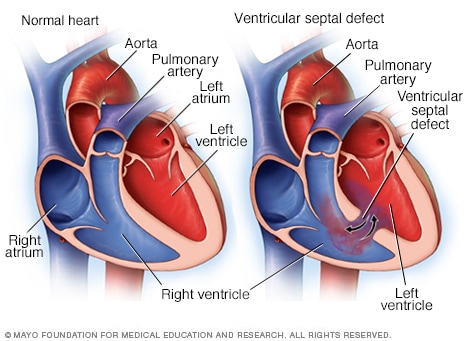
Difetto del setto ventricolare
Un difetto del setto interventricolare è un'apertura anomala (foro) nel cuore che si forma tra le camere di pompaggio inferiori del cuore (ventricoli), come mostrato nel cuore a destra. Ciò consente al sangue ricco di ossigeno e a quello povero di ossigeno di mescolarsi. A sinistra è mostrato un cuore normale.
Tronco arterioso
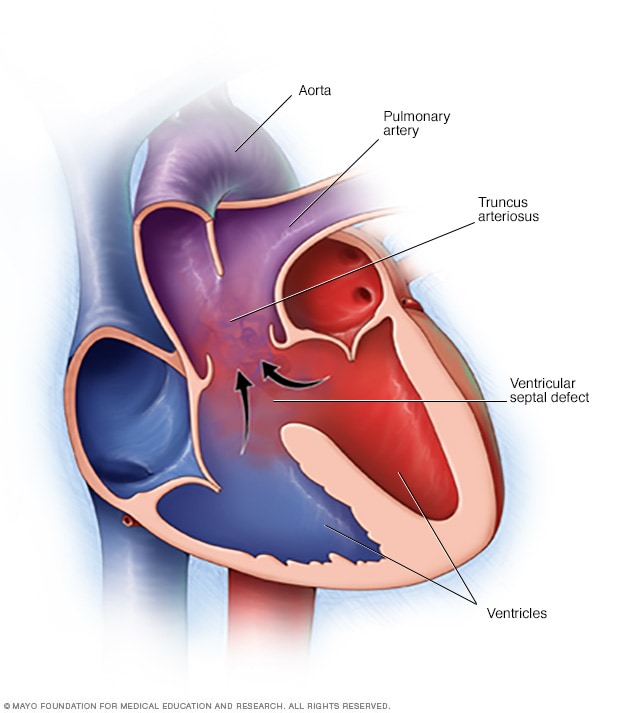
Tronco arterioso
Se tu o il tuo bambino avete un tronco arterioso, invece di due vasi separati, un vaso grande esce dal cuore e c'è un buco nella parete tra le camere cardiache (difetto del setto ventricolare). Il sangue ricco di ossigeno (rosso) e il sangue povero di ossigeno (blu) si mescolano, creando sangue con un apporto di ossigeno insufficiente (viola) al corpo.
Tetralogia di Fallot
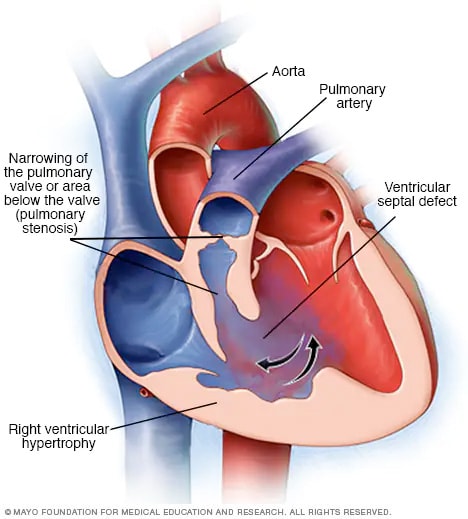
Tetralogia di Fallot
La tetralogia di Fallot è una combinazione di quattro difetti cardiaci congeniti. I quattro difetti sono un difetto del setto ventricolare (VSD), stenosi polmonare, aorta fuori posto e parete ventricolare destra ispessita (ipertrofia ventricolare destra). Di solito provocano una mancanza di sangue ricco di ossigeno che raggiunge il corpo.
ghiandole paratiroidi
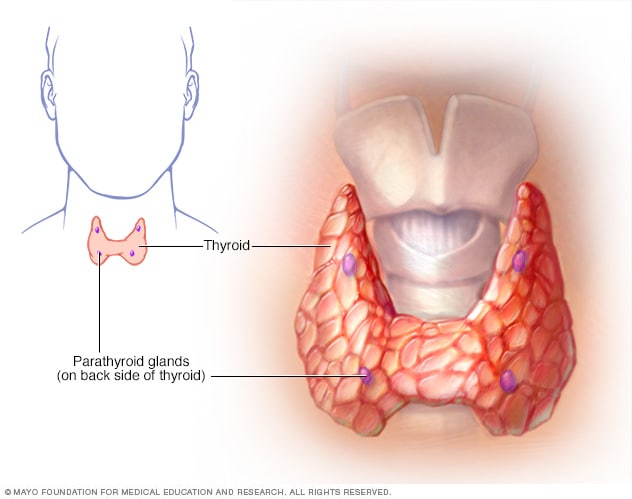
ghiandole paratiroidi
Dietro la tiroide ci sono le ghiandole paratiroidi. Producono l’ormone paratiroideo, che svolge un ruolo nella regolazione dei livelli ematici di calcio e fosforo nel corpo.
Palatoschisi
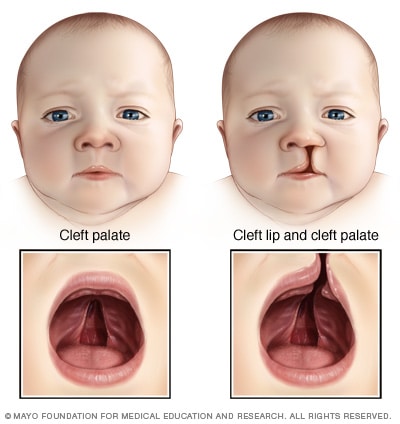
Palatoschisi
Una palatoschisi è un'apertura o una spaccatura nel tetto della bocca che si verifica quando il tessuto non riesce a crescere insieme durante lo sviluppo nell'utero. Una palatoschisi spesso comporta una spaccatura (schisi) nel labbro superiore (labbro leporino), ma può verificarsi senza influenzare il labbro.
Parti del sistema immunitario
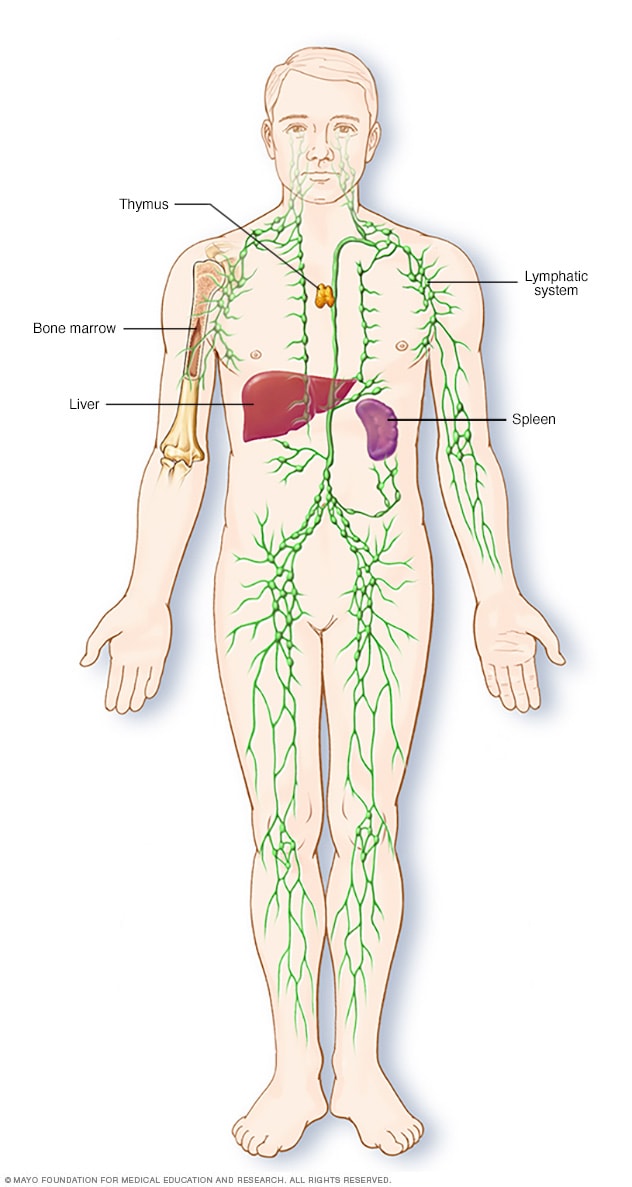
Parti del sistema immunitario
Il sistema linfatico fa parte del sistema immunitario del corpo, che protegge dalle infezioni e dalle malattie. Il sistema linfatico comprende la milza, il timo, i linfonodi e i dotti linfatici, nonché le tonsille e le adenoidi.
Le porzioni del cromosoma 22 delete nella sindrome di DiGeorge (sindrome da delezione 22q11.2) svolgono un ruolo nello sviluppo di numerosi sistemi corporei. Di conseguenza, il disturbo può causare molteplici errori durante lo sviluppo fetale. I problemi comuni che si verificano con la sindrome da delezione 22q11.2 includono:
- Herzfehler. Das 22q11.2-Deletionssyndrom verursacht häufig Herzfehler, die zu einer unzureichenden Versorgung mit sauerstoffreichem Blut führen können. Defekte können beispielsweise ein Loch zwischen den unteren Herzkammern (Ventrikelseptumdefekt) umfassen; nur ein großes Gefäß statt zwei Gefäße, das aus dem Herzen herausführt (Truncus arteriosus); oder eine Kombination von vier abnormen Herzstrukturen (Fallot-Tetralogie).
- Hypoparathyreoidismus. Die vier Nebenschilddrüsen im Nacken regulieren den Kalzium- und Phosphorspiegel im Körper. Das 22q11.2-Deletionssyndrom kann kleinere als normale Nebenschilddrüsen verursachen, die zu wenig Parathormon (PTH) absondern, was zu Hypoparathyreoidismus führt. Dieser Zustand führt zu einem niedrigen Kalziumspiegel und einem hohen Phosphorspiegel im Blut.
- Dysfunktion der Thymusdrüse. In der Thymusdrüse, die sich unter dem Brustbein befindet, reifen T-Zellen – eine Art weißer Blutkörperchen – heran. Reife T-Zellen werden benötigt, um Infektionen zu bekämpfen. Bei Kindern mit 22q11.2-Deletionssyndrom kann die Thymusdrüse klein sein oder fehlen, was zu einer schwachen Immunfunktion und häufigen, schweren Infektionen führt.
- Gaumenspalte. Eine häufige Erkrankung des 22q11.2-Deletionssyndroms ist eine Gaumenspalte – eine Öffnung (Spalte) im Gaumen (Gaumen) – mit oder ohne Lippenspalte. Andere, weniger sichtbare Anomalien des Gaumens, die ebenfalls vorhanden sein können, können das Schlucken erschweren oder bestimmte Laute beim Sprechen erzeugen.
- Ausgeprägte Gesichtszüge. Bei einigen Menschen mit 22q11.2-Deletionssyndrom können eine Reihe besonderer Gesichtszüge vorhanden sein. Dazu können kleine, tief angesetzte Ohren, kurze Augenöffnungen (Lidspalten), geschlossene Augen, ein relativ langes Gesicht, eine vergrößerte Nasenspitze (bauchig) oder eine kurze oder abgeflachte Furche in der Oberlippe gehören.
- Lern-, Verhaltens- und psychische Gesundheitsprobleme. Die Deletion von 22q11.2 kann Probleme mit der Entwicklung und Funktion des Gehirns verursachen, was zu Lern-, sozialen, Entwicklungs- oder Verhaltensproblemen führen kann. Verzögerungen in der Sprachentwicklung von Kleinkindern und Lernschwierigkeiten sind häufig. Manche Kinder entwickeln eine Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS) oder eine Autismus-Spektrum-Störung. Im späteren Leben steigt das Risiko für Depressionen, Angststörungen und andere psychische Störungen.
- Autoimmunerkrankungen. Menschen, die als Kinder aufgrund einer kleinen oder fehlenden Thymusdrüse eine schlechte Immunfunktion hatten, können auch ein erhöhtes Risiko für Autoimmunerkrankungen wie rheumatoide Arthritis oder Morbus Basedow haben.
- Andere Probleme. Eine große Anzahl von Erkrankungen kann mit dem 22q11.2-Deletionssyndrom in Verbindung gebracht werden, wie z. B. Hörbehinderung, schlechtes Sehvermögen, Atemprobleme, schlechte Nierenfunktion und relativ kleine Statur für die eigene Familie.
prevenzione
In alcuni casi, la sindrome di DiGeorge (sindrome da delezione 22q11.2) può essere trasmessa da un genitore affetto a un figlio. Se sei preoccupato per una storia familiare di sindrome da delezione 22q11.2 o se hai già un bambino affetto dalla sindrome, dovresti consultare un medico specializzato in disturbi genetici (genetista) o un consulente genetico per ricevere aiuto nella pianificazione di future gravidanze.
Fonti:
- Nationalbibliothek für Medizin. 22q11.2-Deletionssyndrom. Genetik-Home-Referenz. https://ghr.nlm.nih.gov/condition/22q112-deletion-syndrome#genes. Abgerufen am 25. Mai 2017.
- 22q11.2-Deletionsstörungen (DiGeorge-Syndrom und Velokardiofaziales Syndrom). American Heart Association. http://www.heart.org/HEARTORG/Conditions/CongenitalHeartDefects/AboutCongenitalHeartDefects/22q112-Deletion-Disorders-DiGeorge-Syndrome-and-Velocardiofacial-Syndrome_UCM_309017_Article.jsp#.WSc5wWd1rRE. Abgerufen am 25. Mai 2017.
- DiGeorge-Syndrom (DGS). American Academy of Allergy Asthma & Immunology. http://www.aaaai.org/conditions-and-treatments/primary-immunodeficiency-disease/digeorge-syndrome. Abgerufen am 25. Mai 2017.
- DiGeorge-Syndrom. Merck Manual Professional-Version. https://www.merckmanuals.com/professional/immunology-allergic-disorders/immunodeficiency-disorders/digeorge-syndrome. Abgerufen am 25. Mai 2017.
- Hofstetter AM, et al. Verwendung und Sicherheit von Lebendimpfstoffen beim DiGeorge-Syndrom. Pädiatrie. 2014;133:e946.
- Chromosom 22q11.2-Deletionssyndrom. Nationale Organisation für seltene Erkrankungen. https://rarediseases.org/rare-diseases/chromosome-22q11-2-deletion-syndrome/. Abgerufen am 25. Mai 2017.
- Seroogie CM. DiGeorge (22q11.2-Deletion)-Syndrom: Epidemiologie und Pathogenese. https://www.uptodate.com/contents/search. Abgerufen am 25. Mai 2017.
- Seroogie CM. DiGeorge (22q11.2-Deletion)-Syndrom: Klinische Merkmale und Diagnose. https://www.uptodate.com/contents/search. Abgerufen am 25. Mai 2017.
- Seroogie CM. DiGeorge (22q11.2-Deletion)-Syndrom: Management und Prognose. https://www.uptodate.com/contents/search. Abgerufen am 25. Mai 2017.
- Fragen Sie MayoExpert. 22q11.2-Deletionssyndrom. Rochester, Minnesota: Mayo Foundation for Medical Education and Research; 2016. Zugriff am 10. Mai 2017.
- Babovic-Vuksanovic D (Gutachten). Mayo Clinic, Rochester, Minnesota, 6. Juli 2017.