

Vorwort
Die Kennzeichen der Krebskonzeptualisierung ist ein heuristisches Werkzeug, um die enorme Komplexität von Krebsphänotypen und -genotypen in einen vorläufigen Satz zugrunde liegender Prinzipien zu destillieren. Mit fortschreitendem Wissen über Krebsmechanismen haben sich andere Facetten der Krankheit als potenzielle Verfeinerungen herauskristallisiert. Hierin wird die Aussicht aufgeworfen, dass phänotypische Plastizität und gestörte Differenzierung eine eigenständige charakteristische Fähigkeit sind, und dass nicht-mutationsbedingte epigenetische Reprogrammierung und polymorphe Mikrobiome beide charakteristische ermöglichende Eigenschaften darstellen, die den Erwerb von charakteristischen Fähigkeiten erleichtern. Darüber hinaus können seneszente Zellen unterschiedlicher Herkunft zur Liste der funktionell wichtigen Zelltypen in der Tumormikroumgebung hinzugefügt werden.
Bedeutung
Krebs ist beängstigend in der Breite und dem Umfang seiner Vielfalt, die Genetik, Zell- und Gewebebiologie, Pathologie und Ansprechen auf eine Therapie umfasst. Immer leistungsfähigere experimentelle und rechnerische Werkzeuge und Technologien liefern eine Lawine von „Big Data“ über die unzähligen Manifestationen von Krankheiten, die Krebs umfasst. Das integrative Konzept, das in den Kennzeichen von Krebs verkörpert ist, hilft dabei, diese Komplexität in eine zunehmend logische Wissenschaft zu destillieren, und die vorläufigen neuen Dimensionen, die in dieser Perspektive präsentiert werden, können diesem Bestreben einen Mehrwert verleihen, um die Mechanismen der Krebsentstehung und des bösartigen Fortschreitens besser zu verstehen und Wenden Sie dieses Wissen auf die Krebsmedizin an.
Einführung
Die Hallmarks of Cancer wurden als eine Reihe funktioneller Fähigkeiten vorgeschlagen, die menschliche Zellen auf ihrem Weg von der Normalität zu neoplastischen Wachstumszuständen erwerben, genauer gesagt Fähigkeiten, die für ihre Fähigkeit, bösartige Tumore zu bilden, entscheidend sind. In diesen Artikeln ( 1, 2), Bob Weinberg und ich zählten auf, was wir uns als gemeinsame Gemeinsamkeiten vorstellten, die alle Arten von Krebszellen auf der Ebene des zellulären Phänotyps vereinen. Die Absicht bestand darin, ein konzeptionelles Gerüst bereitzustellen, das es ermöglichen würde, die komplexen Phänotypen verschiedener menschlicher Tumortypen und -varianten in Bezug auf einen gemeinsamen Satz zugrunde liegender zellulärer Parameter zu rationalisieren. Anfänglich sahen wir die komplementäre Einbeziehung von sechs unterschiedlichen Markenfähigkeiten vor und erweiterten diese Zahl später auf acht.
Diese Formulierung wurde von der Erkenntnis beeinflusst, dass sich menschliche Krebserkrankungen als Produkte mehrstufiger Prozesse entwickeln und dass der Erwerb dieser funktionellen Fähigkeiten in gewisser Weise den unterscheidbaren Schritten der Tumorpathogenese zugeordnet werden könnte. Die Vielfalt der malignen Pathogenese, die mehrere Tumortypen und eine zunehmende Fülle von Subtypen umfasst, umfasst verschiedene Aberrationen (und damit erworbene Fähigkeiten und Eigenschaften), die das Ergebnis gewebespezifischer Barrieren sind, die während bestimmter Tumorentstehungswege notwendigerweise umgangen werden. Obwohl wir anerkennen, dass solche spezialisierten Mechanismen hilfreich sein können, haben wir die Bezeichnung der Markenzeichen auf Parameter beschränkt, die einen breiten Einfluss auf das gesamte Spektrum menschlicher Krebsarten haben.
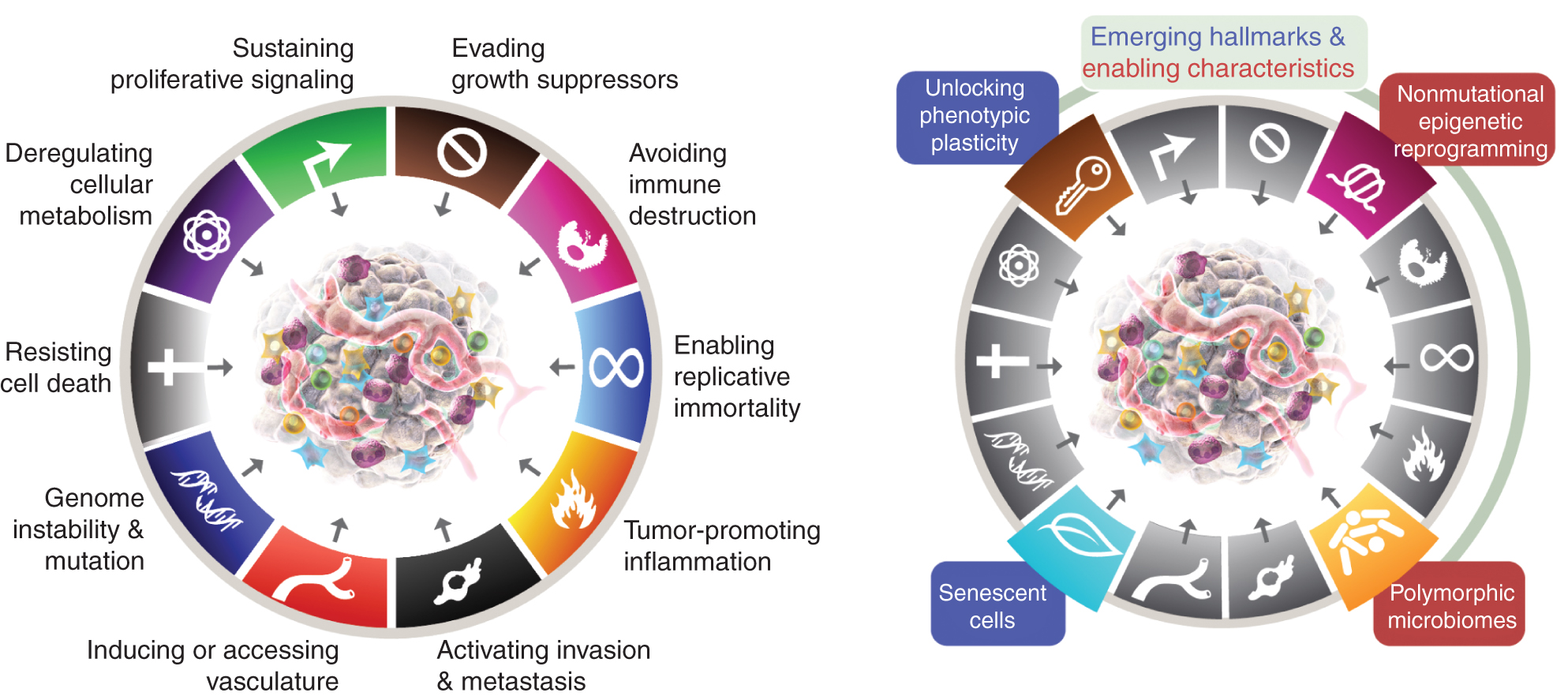
Die acht Markenzeichen umfassen derzeit (Abb.1, links) die erworbenen Fähigkeiten zur Aufrechterhaltung der proliferativen Signalübertragung, zur Vermeidung von Wachstumssuppressoren, zum Widerstand gegen den Zelltod, zur Ermöglichung replikativer Unsterblichkeit, zur Induktion/zum Zugang zu Gefäßen, zur Aktivierung von Invasion und Metastasierung, zur Neuprogrammierung des Zellstoffwechsels und zur Vermeidung der Zerstörung des Immunsystems . In der jüngsten Ausarbeitung dieses Konzepts (2) wurden die Deregulierung des Zellstoffwechsels und die Vermeidung der Zerstörung des Immunsystems als „aufkommende Kennzeichen“ abgegrenzt, aber jetzt, elf Jahre später, ist es offensichtlich, dass sie, ähnlich wie die ursprünglichen sechs, als Kern angesehen werden können Kennzeichen von Krebs und sind als solche in der aktuellen Darstellung enthalten (Abb. 1, links).
Abbildung 1
Kennzeichen von Krebs
Die Kennzeichen von Krebs verkörpern derzeit acht charakteristische Fähigkeiten und zwei unterstützende Eigenschaften. Zusätzlich zu den sechs erworbenen Fähigkeiten – Kennzeichen des Krebses – die im Jahr 2000 (1) vorgeschlagen wurden, wurden die beiden vorläufigen „emergierenden Kennzeichen“ im Jahr 2011 eingeführt (2) – zelluläre Energetik (jetzt allgemeiner als „Umprogrammierung des Zellstoffwechsels“ bezeichnet) und „Vermeidung Immunzerstörung“ – wurden ausreichend validiert, um als Teil des Core-Sets betrachtet zu werden.
Angesichts der wachsenden Erkenntnis, dass Tumore ausreichend vaskularisiert werden können, entweder durch Anschalten der Angiogenese oder durch Kooptierung normaler Gewebegefäße (128), wird dieses Kennzeichen auch allgemeiner definiert als die Fähigkeit, hauptsächlich durch Invasion und Metastasierung Gefäßsysteme zu induzieren oder auf andere Weise zugänglich zu machen, die das Tumorwachstum unterstützen.
Die Fortsetzung von 2011 beinhaltete außerdem „tumorfördernde Entzündung“ als zweites ermöglichendes Merkmal, das die übergreifende „Genominstabilität und -mutation“ ergänzt, die zusammen grundlegend an der Aktivierung der acht charakteristischen (funktionellen) Fähigkeiten beteiligt waren, die für Tumorwachstum und -progression erforderlich sind. Richtig, diese Übersicht enthält zusätzliche vorgeschlagene neue Kennzeichen und ermöglichende Merkmale, darunter „Aufschließen der phänotypischen Plastizität“, „nicht mutationsbedingte epigenetische Reprogrammierung“, „polymorphe Mikrobiome“ und „seneszente Zellen“. Die Markenzeichen der Krebsgrafik wurden von Hanahan und Weinberg (2) übernommen.
Wie wir damals feststellten, können diese charakteristischen Merkmale allein nicht die Komplexität der Krebspathogenese ansprechen, d. h. die genauen molekularen und zellulären Mechanismen, die es den sich entwickelnden präneoplastischen Zellen ermöglichen, diese abweichenden phänotypischen Fähigkeiten zu entwickeln und im Laufe von zu erwerben Tumorentstehung und bösartige Progression.
Dementsprechend haben wir der Diskussion ein weiteres Konzept hinzugefügt, das als „ermöglichende Merkmale“ dargestellt wird, Folgen des abweichenden Zustands der Neoplasie, die Mittel bereitstellen, durch die Krebszellen und Tumore diese funktionellen Merkmale annehmen können. Als solche spiegeln sich die befähigenden Eigenschaften in molekularen und zellulären Mechanismen wider, durch die Markenzeichen erworben werden, und nicht in den oben genannten acht Fähigkeiten selbst. Diese beiden Aktivierungsprozesse waren Genominstabilität und tumorfördernde Entzündung.
Wir haben ferner erkannt, dass die Tumormikroumgebung (TME), die hierin als zusammengesetzt aus heterogenen und interaktiven Populationen von Krebszellen und Krebsstammzellen zusammen mit einer Vielzahl von rekrutierten Stromazelltypen – dem transformierten Parenchym und dem zugehörigen Stroma – definiert ist, heute weithin geschätzt wird eine wesentliche Rolle bei der Tumorentstehung und malignen Progression zu spielen.
Angesichts des anhaltenden Interesses an diesen Formulierungen und unserer anhaltenden Absicht, die laufende Diskussion und Verfeinerung des Hallmarks-Schemas zu fördern, ist es angebracht, eine häufig gestellte Frage zu berücksichtigen: Gibt es zusätzliche Merkmale dieses konzeptionellen Modells, die unter Berücksichtigung der Notwendigkeit, dies sicherzustellen, integriert werden könnten? dass sie im gesamten Spektrum menschlicher Krebsarten breit anwendbar sind? Dementsprechend präsentiere ich mehrere potenzielle neue Kennzeichen und ermöglichende Merkmale, die zu gegebener Zeit als Kernkomponenten der Kennzeichen der Krebskonzeption integriert werden könnten.
Diese Parameter sind „Aufschließen der phänotypischen Plastizität“, „ nicht mutationsbedingte epigenetische Reprogrammierung“, „polymorphe Mikrobiome“ und „seneszente Zellen“ (Abb. 1, Rechts). Wichtig ist, dass die Beispiele, die zur Untermauerung dieser Thesen präsentiert werden, illustrativ, aber keinesfalls umfassend sind, da es eine wachsende und zunehmend überzeugende Menge an veröffentlichten Beweisen zur Untermauerung jeder Vignette gibt.
Phänotypische Plastizität erschließen
Während der Organogenese wird die Entwicklung, Bestimmung und Organisation von Zellen zu Geweben, um homöostatische Funktionen zu übernehmen, von einer terminalen Differenzierung begleitet, wobei Vorläuferzellen – manchmal unwiderruflich – aufhören zu wachsen, wenn diese Prozesse kulminieren. Als solches ist das Endergebnis der zellulären Differenzierung in den meisten Fällen antiproliferativ und bildet eine klare Barriere für die fortgesetzte Proliferation, die für Neoplasien notwendig ist.
Es gibt zunehmend Hinweise darauf, dass das Aufschließen der normalerweise eingeschränkten Fähigkeit zur phänotypischen Plastizität, um den Zustand der terminalen Differenzierung zu umgehen oder ihm zu entkommen, eine entscheidende Komponente der Krebspathogenese ist (3). Diese Plastizität kann in mehreren Erscheinungsformen wirken (Abb. 2). So können naszierende Krebszellen, die aus einer normalen Zelle stammen, die sich auf einem Weg entwickelt hat, der sich einem vollständig differenzierten Zustand nähert oder diesen annimmt, ihren Kurs umkehren, indem sie zurück zu Vorläufer-ähnlichen Zellzuständen dedifferenzieren.
Umgekehrt können neoplastische Zellen, die aus einer Vorläuferzelle entstehen, die dazu bestimmt ist, einem Weg zu folgen, der zur Differenzierung im Endstadium führt, den Prozess kurzschließen und die expandierenden Krebszellen in einem teilweise differenzierten, Vorläufer-ähnlichen Zustand halten. Alternativ kann eine Transdifferenzierung stattfinden, bei der Zellen, die ursprünglich auf einen Differenzierungsweg festgelegt wurden, zu einem völlig anderen Entwicklungsprogramm wechseln und dadurch gewebespezifische Merkmale erwerben, die nicht durch ihre normalen Ursprungszellen vorherbestimmt wurden.
Die folgenden Beispiele stützen das Argument, dass unterschiedliche Formen der zellulären Plastizität phänotypische Plastizität erschließen. Links ist die phänotypische Plastizität wohl eine erworbene charakteristische Fähigkeit, die verschiedene Störungen der Zelldifferenzierung ermöglicht, einschließlich (i) Dedifferenzierung von reifen zu Vorläuferzuständen, (ii) blockierte (terminale) Differenzierung von Vorläuferzellzuständen und (iii) Transdifferenzierung in andere Zellen Linien. Rechts sind drei prominente Modi der gestörten Differenzierung dargestellt, die integraler Bestandteil der Krebspathogenese sind.
Durch unterschiedliche Verfälschung der normalen Differenzierung von Vorläuferzellen zu reifen Zellen in Entwicklungslinien wird die Tumorentstehung und maligne Progression, die aus Ursprungszellen in solchen Wegen hervorgeht, erleichtert. Die Markenzeichen der Krebsgrafik wurden von Hanahan und Weinberg (2) übernommen.
Abbildung 2

Phänotypische Plastizität
Entdifferenzierung
Die Kolonkarzinogenese ist ein Beispiel für eine gestörte Differenzierung, da für beginnende Krebszellen eine teleologische Notwendigkeit besteht, dem Förderband der terminalen Differenzierung und Exfoliation zu entkommen, was im Prinzip durch eine Dedifferenzierung von noch nicht endgültig terminal differenzierten Kolonepithelzellen oder durch eine blockierte Differenzierung erfolgen könnte von Vorläufer-/Stammzellen in den Krypten, die diese differenzierenden Zellen hervorbringen. Sowohl differenzierte Zellen als auch Stammzellen wurden als Ursprungszellen für Dickdarmkrebs in Verbindung gebracht ( 4–6).
Zwei Entwicklungstranskriptionsfaktoren (TF), das Homöobox-Protein HOXA5 und SMAD4, das letztere, das an der BMP-Signalübertragung beteiligt ist, werden in differenzierenden Dickdarmepithelzellen stark exprimiert und gehen typischerweise in fortgeschrittenen Dickdarmkarzinomen verloren, die charakteristischerweise Marker von Stamm- und Vorläuferzellen exprimieren. Funktionelle Störungen in Mausmodellen haben gezeigt, dass die erzwungene Expression von HOXA5 in Dickdarmkrebszellen Differenzierungsmarker wiederherstellt, Stammzellphänotypen unterdrückt und Invasion und Metastasierung beeinträchtigt, was eine Begründung für seine charakteristische Herunterregulierung liefert ( 7, 8).
Im Gegensatz dazu erzwingt SMAD4 sowohl die Differenzierung als auch die Unterdrückung der Proliferation, die durch die onkogene WNT-Signalgebung angetrieben wird, was durch den manipulierten Verlust der SMAD4-Expression aufgedeckt wird, was eine Erklärung für seinen Expressionsverlust liefert, um eine Dedifferenzierung und anschließend eine WNT-gesteuerte Hyperproliferation zu ermöglichen ( 5 ).
Bemerkenswerterweise ist der Verlust dieser beiden „Differenzierungsunterdrücker“ mit der daraus resultierenden Dedifferenzierung mit dem Erwerb anderer Kennzeichenfähigkeiten verbunden, ebenso wie andere Kennzeichen-induzierende Regulatoren, was die strenge Definition dieses vorläufigen Kennzeichens als trennbar und unabhängig erschwert.
Eine weitere Beweisführung betrifft die unterdrückte Expression des MITF -Hauptregulators der Melanozytendifferenzierung, der offensichtlich an der Genese aggressiver Formen des malignen Melanoms beteiligt ist. Der Verlust dieses Entwicklungs-TF ist mit der Reaktivierung von Neuralleisten-Vorläufergenen und der Herunterregulierung von Genen verbunden, die vollständig differenzierte Melanozyten charakterisieren. Das Wiedererscheinen der Neuralleisten-Gene weist darauf hin, dass diese Zellen in den Vorläuferzustand zurückkehren, aus dem Melanozyten entwicklungsbedingt entstehen.
Darüber hinaus etablierte eine Abstammungsverfolgungsstudie von BRAF -induzierten Melanomen reife pigmentierte Melanozyten als Ursprungszellen, die im Verlauf der Tumorentstehung einer Dedifferenzierung unterliegen ( 9 ). Bemerkenswert ist die Mutante BRAFOnkogen, das in mehr als der Hälfte der kutanen Melanome gefunden wird, induziert eine Hyperproliferation, die vorangeht und daher mechanistisch von der nachfolgenden Dedifferenzierung getrennt werden kann, die aus der Herunterregulierung von MITF entsteht.
Eine andere Studie implizierte funktionell die Hochregulierung des Entwicklungs-TF ATF2 , dessen charakteristische Expression in Maus- und menschlichen Melanomen indirekt MITF1 unterdrückt , einhergehend mit einer malignen Progression der folglich dedifferenzierten Melanomzellen ( 10 ). Umgekehrt führt die Expression von mutanten Formen von ATF2 , die MITF nicht unterdrücken können, in Melanomen zu gut differenzierten Melanomen ( 11 ).
Darüber hinaus hat eine kürzlich durchgeführte Studie ( 12 ) die Dedifferenzierung der Abstammungslinie mit der malignen Progression von Inselzellneoplasien der Bauchspeicheldrüse zu metastasenanfälligen Karzinomen in Verbindung gebracht; diese neuroendokrinen Zellen und abgeleiteten Tumore entstehen aus einer Entwicklungslinie, die sich von derjenigen unterscheidet, die die weitaus größere Anzahl benachbarter Zellen erzeugt, die das exokrine und Pankreas und die daraus entstehenden duktalen Adenokarzinome bilden.
Bemerkenswerterweise wurde der mehrstufige Differenzierungsweg von Insel-Vorläuferzellen zu reifen β-Zellen gründlich charakterisiert ( 13). Vergleichendes Transkriptom-Profiling zeigt, dass Adenom-ähnliche Inseltumoren unreifen, aber differenzierten insulinproduzierenden β-Zellen am ähnlichsten sind, während die invasiven Karzinome embryonalen Inselzellvorläufern am ähnlichsten sind. Die Progression zu schlecht differenzierten Karzinomen beinhaltet einen ersten Schritt der Dedifferenzierung, der im Vergleich zu den gut differenzierten Adenomen, die beide eher später auftreten, zunächst keine verstärkte Proliferation oder verringerte Apoptose beinhaltet.
Somit wird der diskrete Schritt der Dedifferenzierung nicht durch beobachtbare Veränderungen in den charakteristischen Merkmalen der anhaltenden Proliferation und Resistenz gegen Apoptose angetrieben. Vielmehr ist die Hochregulierung einer miRNA, die zuvor an der Spezifizierung des Inselvorläuferzustands beteiligt war, einer, der während der terminalen Differenzierung von β-Zellen herunterreguliert wird, 12).
Blockierte Differenzierung
Während die obigen Beispiele veranschaulichen, wie die Unterdrückung der Differenzierungsfaktorexpression die Tumorentstehung erleichtern kann, indem es besser differenzierten Zellen ermöglicht wird, sich zu Vorläufern zu dedifferenzieren, können unvollständig differenzierte Vorläuferzellen in anderen Fällen regulatorische Veränderungen erleiden, die aktiv ihr weiteres Vordringen in vollständig differenzierte, typischerweise nicht proliferative Zustände blockieren .
Es ist seit langem dokumentiert, dass die akute Promyelozytenleukämie (APL) aus einer chromosomalen Translokation resultiert, die den PML -Locus mit dem Gen fusioniert, das den Retinsäure-α-Kernrezeptor (RARα) kodiert. Myeloische Vorläuferzellen, die solche Translokationen tragen, sind offensichtlich nicht in der Lage, ihre übliche terminale Differenzierung zu Granulozyten fortzusetzen, was zu Zellen führt, die in einem proliferativen, promyelozytenartigen Vorläuferstadium gefangen sind ( 14 ).
Der Konzeptnachweis dieses Schemas stammt aus der Behandlung von kultivierten APL-Zellen, Mausmodellen dieser Krankheit sowie von betroffenen Patienten mit Retinsäure, dem Liganden von RARα; Diese therapeutische Behandlung bewirkt, dass sich die neoplastischen APL-Zellen zu scheinbar reifen, nicht proliferierenden Granulozyten differenzieren, wodurch ihre fortschreitende proliferative Expansion kurzgeschlossen wird (14–16 ).
Eine Variation dieses Themas betrifft eine andere Form der akuten myeloischen Leukämie, diese trägt die t(8;21)-Translokation, die das AML1-ETO-Fusionsprotein produziert. Dieses Protein kann allein myeloische Vorläufer transformieren, zumindest teilweise, indem es ihre Differenzierung blockiert. Die therapeutische Intervention in Mausmodellen und bei Patienten mit einem pharmakologischen Inhibitor einer Chromatin-modifizierenden Histon-Deacetylase (HDAC) bewirkt, dass die myeloischen Leukämiezellen ihre Differenzierung in Zellen mit einer reiferen myeloischen Zellmorphologie wieder aufnehmen. Einhergehend mit dieser Reaktion ist eine Verringerung der Proliferationsfähigkeit, wodurch das Fortschreiten dieser Leukämie beeinträchtigt wird ( 17, 18 ).
Ein drittes Beispiel betrifft beim Melanom einen Entwicklungs-TF, SOX10 , der normalerweise während der Melanozytendifferenzierung herunterreguliert wird. Funktionsgewinn- und -verluststudien in einem Zebrafischmodell von BRAF – induzierten Melanomen haben gezeigt, dass eine anormal aufrechterhaltene Expression von SOX10 die Differenzierung von neuralen Vorläuferzellen zu Melanozyten blockiert, was die Bildung von BRAF – gesteuerten Melanomen ermöglicht ( 19 ).
Weitere Beispiele für Differenzierungsmodulatoren sind der Metabolit Alpha-Ketoglutarat (αKG), ein notwendiger Cofaktor für eine Reihe von Chromatin-modifizierenden Enzymen, der nachweislich an der Stimulierung bestimmter differenzierter Zellzustände beteiligt ist. Bei Bauchspeicheldrüsenkrebs stimuliert der Tumorsuppressor p53 die Produktion von αKG und die Aufrechterhaltung eines besser differenzierten Zellzustands, während ein prototypischer Verlust der p53-Funktion zu einer Verringerung der αKG-Spiegel und einer daraus resultierenden Dedifferenzierung führt, die mit einer malignen Progression verbunden ist ( 20 ).
Bei einer Form von Leberkrebs ist die Mutation eines Isocitrat-Dehydrogenase-Gens ( IDH1/2) führt nicht zur Produktion von differenzierungsinduzierendem αKG, sondern eher zu einem verwandten „Onkometaboliten“, D-2-Hydroxygluterat (D2HG), von dem gezeigt wurde, dass es die Hepatozytendifferenzierung von Lebervorläuferzellen durch D2HG-vermittelte Repression eines Hauptregulators von blockiert Hepatozytendifferenzierung und Ruhe, HNF4a.
Die D2HG-vermittelte Unterdrückung der HNF4a-Funktion löst eine proliferative Expansion der Hepatozyten-Vorläuferzellen in der Leber aus, die bei anschließender mutationsbedingter Aktivierung des KRAS – Onkogens, das die maligne Progression zum Cholangiokarzinom der Leber vorantreibt, für eine onkogene Transformation anfällig werden ( 21). Die Mutante IDH1/2 und ihr Onkometabolit D2HG wirken auch bei einer Vielzahl von myeloischen und anderen soliden Tumortypen, bei denen D2HG αKG-abhängige Dioxygenasen hemmt, die für Histon- und DNA-Methylierungsereignisse erforderlich sind, die Veränderungen in der Chromatinstruktur während der Differenzierung der Entwicklungslinie vermitteln, wodurch der Beginn eingefroren wird Krebszellen in einem Vorläuferzustand ( 22, 23 ).
Ein zusätzliches, verwandtes Konzept ist die „umgangene Differenzierung“, bei der teilweise oder undifferenzierte Vorläufer-/Stammzellen den Zellzyklus verlassen und in Schutznischen schlummern, mit dem Potenzial, die proliferative Expansion ( 24 ) neu zu initiieren, wenn auch immer noch mit dem selektiven Druck zu stören ihre programmierte Differenzierung auf die eine oder andere Weise.
Transdifferenzierung
Das Konzept der Transdifferenzierung wird seit langem von Pathologen in Form von Gewebemetaplasie erkannt, bei der Zellen eines bestimmten differenzierten Phänotyps ihre Morphologie deutlich ändern, um klar als Elemente eines anderen Gewebes erkennbar zu werden, wofür ein prominentes Beispiel der Barrett-Ösophagus ist, wo chronische Entzündungen auftreten des mehrschichtigen Plattenepithels des Ösophagus induziert eine Transdifferenzierung in ein für den Darm charakteristisches einfaches Zylinderepithel, wodurch die spätere Entwicklung von Adenokarzinomen erleichtert wird und nicht die aus diesem Plattenepithel zu erwartenden Plattenepithelkarzinome (3).
Jetzt offenbaren molekulare Determinanten Mechanismen der Transdifferenzierung bei verschiedenen Krebsarten, sowohl für Fälle, in denen eine grobe Gewebemetaplasie offensichtlich ist, als auch für andere, in denen sie etwas subtiler ist, wie die folgenden Beispiele veranschaulichen.
Ein aufschlussreicher Fall für die Transdifferenzierung als diskretes Ereignis bei der Tumorentstehung betrifft das duktale Adenokarzinom des Pankreas (PDAC), bei dem eine der beteiligten Ursprungszellen, die Bauchspeicheldrüsen-Azinuszelle, während der Initiierung der neoplastischen Entwicklung in einen duktalen Zellphänotyp transdifferenziert werden kann. Zwei TFs – PTF1a und MIST1 – steuern über ihre Expression im Zusammenhang mit sich selbst erhaltenden „Feed-Forward“-Regulationsschleifen die Spezifikation und Aufrechterhaltung des differenzierten Azinuszellenzustands der Bauchspeicheldrüse ( 25 ).
Beide dieser TFs werden häufig während der neoplastischen Entwicklung und der malignen Progression von humanen und Maus-PDAC herunterreguliert. Funktionelle genetische Studien an Mäusen und kultivierten menschlichen PDAC-Zellen haben gezeigt, dass die experimentell erzwungene Expression von PTF1abeeinträchtigt die KRAS -induzierte Transdifferenzierung und Proliferation und kann auch die Redifferenzierung von bereits neoplastischen Zellen in einen ruhenden Azinuszellphänotyp erzwingen ( 26 ).
Umgekehrt löst die Unterdrückung der PTF1a -Expression eine Azinus-zu-Duktus-Metaplasie aus, nämlich Transdifferenzierung, und sensibilisiert dadurch die Duktus-ähnlichen Zellen für die onkogene KRAS – Transformation, was die nachfolgende Entwicklung von invasivem PDAC beschleunigt ( 27 ). Ebenso erzwungene Expression von MIST1 in KRAS-exprimierende Bauchspeicheldrüse blockiert auch die Transdifferenzierung und beeinträchtigt die Initiierung der Pankreas-Tumorgenese, die sonst durch die Bildung von prämalignen gangartigen (PanIN) Läsionen erleichtert wird, während die genetische Deletion von MIST1 ihre Bildung und die Initiierung der KRAS – gesteuerten neoplastischen Progression verstärkt ( 28 ).
Der Verlust entweder der PTF1- oder der MIST1- Expression während der Tumorgenese ist mit einer erhöhten Expression eines anderen entwicklungsregulatorischen TF, SOX9 , verbunden, der normalerweise bei der Spezifikation von Duktuszellen wirksam ist ( 27, 28 ). Erzwungene Hochregulierung von SOX9 , wodurch die Notwendigkeit einer Herunterregulierung von PTF1a vermieden wird undEs wurde auch gezeigt, dass MIST1 die Transdifferenzierung von Azinuszellen in einen duktalen Zellphänotyp stimuliert, der für KRAS – induzierte Neoplasien empfindlich ist ( 29 ), was SOX9 als einen funktionellen Schlüsseleffektor ihrer Herunterregulierung bei der Genese von menschlichem PDAC impliziert.
Somit können drei TFs, die die Differenzierung der Bauchspeicheldrüse regulieren, auf verschiedene Weise verändert werden, um einen transdifferenzierten Zustand zu induzieren, der – im Zusammenhang mit der mutationsbedingten Aktivierung von KRAS – die onkogene Transformation und die Initiierung der Tumorentstehung und des malignen Fortschreitens erleichtert.
Weitere Mitglieder der SOX-Familie von Chromatin-assoziierten regulatorischen Faktoren sind einerseits weitgehend sowohl mit der Spezifikation des Zellschicksals als auch mit dem Wechsel der Abstammungslinie in der Entwicklung ( 30 ) und andererseits mit mehreren tumorassoziierten Phänotypen ( 31 ) assoziiert). Ein weiteres hervorstechendes Beispiel einer SOX-vermittelten Transdifferenzierung beinhaltet einen Mechanismus der therapeutischen Resistenz bei Prostatakarzinomen.
In diesem Fall ist der Verlust der RB- und p53-Tumorsuppressoren – deren Fehlen charakteristisch für neuroendokrine Tumoren ist – als Reaktion auf eine Antiandrogentherapie notwendig, aber nicht ausreichend für die häufig beobachtete Umwandlung von gut differenzierten Prostatakrebszellen in eingedrungene Karzinomzellen Differenzierungslinie mit molekularen und histologischen Merkmalen von neuroendokrinen Zellen, die insbesondere den Androgenrezeptor nicht exprimieren. Zusätzlich zum Verlust von RB und p53 erfordert die erworbene Resistenz gegen eine Antiandrogentherapie eine hochregulierte Expression von SOX2entwicklungsregulatorisches Gen, das nachweislich dazu beiträgt, die Transdifferenzierung der therapieresponsiven Adenokarzinomzellen in Derivate zu induzieren, die sich in einem neuroendokrinen Zellzustand befinden, der gegenüber der Therapie refraktär ist (32).
Ein drittes Beispiel zeigt auch die Transdifferenzierung als eine Strategie, die von Karzinomzellen angewandt wird, um eine Eliminierung durch eine linienspezifische Therapie zu vermeiden, in diesem Fall mit Basalzellkarzinomen (BCC) der Haut, die mit einem pharmakologischen Inhibitor des Hedgehog-Smoothened (HH/SMO ) onkogener Signalweg, von dem bekannt ist, dass er das neoplastische Wachstum dieser Zellen antreibt ( 33 ).
Arzneimittelresistente Krebszellen wechseln über breite epigenetische Verschiebungen in bestimmten Chromatindomänen und die veränderte Zugänglichkeit von zwei Superenhancern zu einem entwicklungsbedingt verwandten, aber unterschiedlichen Zelltyp. Der neu gewonnene phänotypische Zustand der BCC-Zellen ermöglicht es ihnen, die Expression des onkogenen WNT -Signalwegs aufrechtzuerhalten, was wiederum die Unabhängigkeit von dem medikamentenunterdrückten HH/SMO verleihtSignalweg ( 34 ).
Wie aufgrund dieser Transdifferenzierung zu erwarten war, verschiebt sich das Transkriptom der Krebszellen von einer Gensignatur, die die beteiligte Ursprungszelle von BCCs widerspiegelt, nämlich die Stammzellen der Haarfollikelwölbung, zu einer Signatur, die auf die basalen Stammzellen hinweist, die die BZK bevölkern interfollikuläre Epidermis. Eine solche Transdifferenzierung zur Ermöglichung einer Arzneimittelresistenz wird zunehmend bei verschiedenen Krebsformen dokumentiert ( 35 ).
Die Plastizität der Entwicklungslinie scheint auch bei den wichtigsten Subtypen von Lungenkarzinomen vorherrschend zu sein, d. h. bei neuroendokrinen Karzinomen [kleinzelliger Lungenkrebs (SCLC)] und Adenokarzinomen + Plattenepithelkarzinomen [kollektiv nicht-kleinzelliger Lungenkrebs (NSCLC)]. Die Einzelzell-RNA-Sequenzierung hat eine bemerkenswert dynamische und heterogene Umwandlung zwischen diesen Subtypen sowie deutliche Variationen davon während der Stadien der Lungentumorentstehung, der anschließenden malignen Progression und des Ansprechens auf die Therapie offenbart ( 36-38).
Anstelle der einfachen Konzeptualisierung eines reinen klonalen Wechsels von einer Linie in eine andere zeichnen diese Studien daher ein viel komplexeres Bild von sich dynamisch ineinander umwandelnden Subpopulationen von Krebszellen, die Merkmale mehrerer Entwicklungslinien und Differenzierungsstadien aufweisen, eine ernüchternde Erkenntnis in dieser Hinsicht zum abstammungsbasierten therapeutischen Targeting von menschlichem Lungenkrebs. Regulatorische Determinanten dieser dynamischen phänotypischen Plastizität werden allmählich identifiziert ( 37, 39, 40 ).
Zusammenfassung
Die drei oben beschriebenen Klassen von Mechanismen heben selektive Regulatoren der zellulären Plastizität hervor, die – zumindest teilweise – von onkogenen Kerntreibern und anderen charakteristischen Fähigkeiten trennbar sind. Über diese Beispiele hinaus gibt es eine beträchtliche Menge an Beweisen, die viele Formen von Krebs mit einer gestörten Differenzierung in Verbindung bringen, die mit dem Erwerb von Transkriptomsignaturen und anderen Phänotypen einhergeht – zum Beispiel histologische Morphologie –, die mit Vorläufer- oder Stammzellstadien assoziiert sind, die in den entsprechenden normalen Geweben beobachtet werden. Herkunft oder in anderen entfernter verwandten Zelltypen und Abstammungslinien ( 41–43).
Als solche scheinen diese drei Unterklassen der phänotypischen Plastizität – Dedifferenzierung reifer Zellen zurück in Vorläuferzustände, blockierte Differenzierung zum Einfrieren sich entwickelnder Zellen in Vorläufer-/Stammzellzuständen und Transdifferenzierung zu alternativen Zelllinien – bei mehreren Krebsarten während des Primärtumors wirksam zu sein Entstehung, bösartige Progression und/oder Ansprechen auf die Therapie.
Es gibt jedoch zwei konzeptionelle Überlegungen. Erstens sind Dedifferenzierung und blockierte Differenzierung wahrscheinlich miteinander verflochten, da sie bei vielen Tumorarten, bei denen die Ursprungszelle – differenzierte Zelle oder Vorläufer-/Stammzelle – entweder unbekannt oder alternativ beteiligt ist, nicht unterscheidbar sind. Zweitens ist der Erwerb oder Erhalt von Vorläuferzellphänotypen und der Verlust differenzierter Merkmale in den meisten Fällen eine ungenaue Widerspiegelung des normalen Entwicklungsstadiums, Eintauchen in ein Milieu anderer charakteristischer Veränderungen in der Krebszelle, die in sich natürlich entwickelnden Zellen nicht vorhanden sind.
Darüber hinaus beinhaltet eine weitere Form der phänotypischen Plastizität die Zellalterung, die unten allgemeiner diskutiert wird, wobei Krebszellen, die zu einer scheinbar irreversiblen Alterung veranlasst werden, stattdessen in der Lage sind, zu entkommen und die proliferative Expansion fortzusetzen (44 ). Schließlich ist die zelluläre Plastizität, wie bei anderen charakteristischen Fähigkeiten, keine neue Erfindung oder Verirrung von Krebszellen, sondern eher die Korruption latenter, aber aktivierbarer Fähigkeiten, die verschiedene normale Zellen verwenden, um die Homöostase, Reparatur und Regeneration zu unterstützen ( 45 ).
Insgesamt ermutigen diese anschaulichen Beispiele zur Überlegung, dass die Freischaltung der zellulären Plastizität, um verschiedene Formen der gestörten Differenzierung zu ermöglichen, eine eigenständige charakteristische Fähigkeit darstellt, die sich in Regulation und zellulärem Phänotyp von den gut validierten Kernmerkmalen von Krebs unterscheidet ( Abb. 2 ).
Epigenetische Reprogrammierung ohne Mutation
Die ermöglichende Eigenschaft der Instabilität und Mutation des Genoms (DNA) ist eine grundlegende Komponente der Krebsentstehung und -pathogenese. Gegenwärtig katalogisieren mehrere internationale Konsortien Mutationen im gesamten Genom menschlicher Krebszellen, und zwar bei praktisch jeder Art von menschlichem Krebs, in verschiedenen Stadien des bösartigen Fortschreitens, einschließlich metastatischer Läsionen, und während der Entwicklung einer adaptiven Therapieresistenz. Ein Ergebnis ist die inzwischen weit verbreitete Erkenntnis, dass Mutationen in Genen, die die Chromatinarchitektur organisieren, modulieren und aufrechterhalten und dadurch die Genexpression global regulieren, zunehmend entdeckt und funktionell mit Krebsmerkmalen in Verbindung gebracht werden ( 46–48 ).
Darüber hinaus gibt es Argumente für eine andere scheinbar unabhängige Form der Genom-Reprogrammierung, die rein epigenetisch regulierte Änderungen in der Genexpression beinhaltet, eine, die als „nicht-mutationsbedingte epigenetische Reprogrammierung“ bezeichnet werden könnte ( Abb. 3 ). Tatsächlich wurde die These einer mutationslosen Krebsevolution und einer rein epigenetischen Programmierung charakteristischer Krebsphänotypen vor fast einem Jahrzehnt erhoben ( 49 ) und wird zunehmend diskutiert ( 46, 50–52 ).
Abbildung 3

Epigenetische Reprogrammierung ohne Mutation
Ähnlich wie während der Embryogenese und der Gewebedifferenzierung und Homöostase sprechen immer mehr Beweise dafür, dass instrumentelle Genregulationsschaltkreise und -netzwerke in Tumoren von einer Fülle von korrumpierten und kooptierten Mechanismen gesteuert werden können, die unabhängig von Genominstabilität und Genmutation sind. Die Markenzeichen der Krebsgrafik wurden von Hanahan und Weinberg (2) übernommen.
Das Konzept der nicht-mutationsbedingten epigenetischen Regulation der Genexpression ist natürlich als der zentrale Mechanismus, der die embryonale Entwicklung, Differenzierung und Organogenese vermittelt, gut etabliert ( 53–55 ). Beim Erwachsenen zum Beispiel beinhaltet das Langzeitgedächtnis Veränderungen in der Gen- und Histonmodifikation, in der Chromatinstruktur und in der Auslösung von Genexpressionsschaltern, die über die Zeit durch positive und negative Rückkopplungsschleifen stabil aufrechterhalten werden ( 56, 57 ). Zunehmende Beweise stützen die These, dass analoge epigenetische Veränderungen zum Erwerb charakteristischer Fähigkeiten während der Tumorentwicklung und bösartigen Progression beitragen können. Zur Unterstützung dieser Hypothese werden im Folgenden einige Beispiele vorgestellt.
Mikroumgebungsmechanismen der epigenetischen Reprogrammierung
Wenn nicht allein durch onkogene Mutationen, wie wird dann das Krebszellgenom umprogrammiert? Eine wachsende Zahl von Beweisen deutet darauf hin, dass die abweichenden physikalischen Eigenschaften der Tumormikroumgebung breite Veränderungen im Epigenom verursachen können, von denen Veränderungen, die für die phänotypische Auswahl von charakteristischen Fähigkeiten vorteilhaft sind, zu einem klonalen Auswachsen von Krebszellen mit verbesserter Eignung für die proliferative Expansion führen können.
Ein gemeinsames Merkmal von Tumoren (oder Regionen innerhalb von Tumoren) ist Hypoxie als Folge einer unzureichenden Vaskularisierung. Hypoxie zum Beispiel reduziert die Aktivität der TET-Demethylasen, was zu erheblichen Veränderungen im Methylom, insbesondere zu einer Hypermethylierung führt ( 58). Eine unzureichende Vaskularisierung schränkt wahrscheinlich auch die Bioverfügbarkeit kritischer, durch das Blut übertragener Nährstoffe ein, und Nährstoffentzug verändert beispielsweise nachweislich die Translationskontrolle und verstärkt folglich den malignen Phänotyp von Brustkrebszellen ( 59 ).
Ein überzeugendes Beispiel für Hypoxie-vermittelte epigenetische Regulation ist eine Form des ausnahmslos tödlichen pädiatrischen Ependymoms. Wie vielen embryonalen und pädiatrischen Tumoren fehlen dieser Form wiederkehrende Mutationen, insbesondere ein Mangel an Treibermutationen in Onkogenen und Tumorsuppressoren. Vielmehr wird das anomale Wachstum dieser Krebszellen nachweislich durch ein durch Hypoxie induziertes genregulatorisches Programm gesteuert ( 60, 61 ). Bemerkenswerterweise befindet sich die mutmaßliche Ursprungszelle dieses Krebses in einem hypoxischen Kompartiment und sensibilisiert wahrscheinlich darin befindliche Zellen für die Initiierung der Tumorentstehung durch noch unbekannte Cofaktoren.
Ein weiterer überzeugender Beweis für eine mikroumgebungsvermittelte epigenetische Regulation betrifft die invasive Wachstumsfähigkeit von Krebszellen. Ein klassisches Beispiel ist die reversible Induktion der Invasivität von Krebszellen an den Rändern vieler solider Tumore, orchestriert durch das entwicklungsregulatorische Programm, das als Epithel-zu-Mesenchym-Übergang (EMT; Lit. 62–64 ) bekannt ist. Bemerkenswerterweise wurde kürzlich gezeigt, dass ein Hauptregulator der EMT, ZEB1 , die Expression einer Histonmethyltransferase, SETD1B , induziert, die wiederum die ZEB1- Expression in einer positiven Rückkopplungsschleife aufrechterhält, die den (invasiven) regulatorischen Zustand der EMT aufrechterhält ( 65).
Eine frühere Studie dokumentierte in ähnlicher Weise, dass die Induktion von EMT durch hochregulierte Expression eines verwandten TF, SNAIL1 , deutliche Veränderungen in der Chromatinlandschaft infolge der Induktion einer Reihe von Chromatinmodifikatoren verursachte, deren Aktivität nachweislich für die Aufrechterhaltung des phänotypischen Zustands notwendig war ( 66 ). Darüber hinaus kann eine Reihe von Zuständen und Faktoren, denen Krebszellen an den Rändern von Tumoren ausgesetzt sind, einschließlich Hypoxie und von Stromazellen sezernierter Zytokine, offensichtlich die EMT und damit die Invasivität induzieren ( 67, 68 ).
Ein markantes Beispiel für die Programmierung der Invasivität durch die Mikroumgebung, die angeblich nichts mit dem EMT-Programm zu tun hat, beinhaltet die autokrine Aktivierung eines neuronalen Signalschaltkreises, der sekretiertes Glutamat und seinen Rezeptor NMDAR umfasst ( 69, 70). Bemerkenswerterweise hat die prototypische Steifheit vieler solider Tumore, die in umfangreichen Veränderungen der extrazellulären Matrix (ECM) verkörpert ist, die die Zellen darin umhüllt, weitreichende Auswirkungen auf die invasiven und anderen phänotypischen Eigenschaften von Krebszellen.
Verglichen mit der normalen Gewebe-EZM, aus der Tumore stammen, ist die Tumor-EZM typischerweise durch erhöhte Vernetzung und Dichte, enzymatische Modifikationen und veränderte molekulare Zusammensetzung gekennzeichnet, die kollektiv – teilweise über Integrinrezeptoren für ECM-Motive – steifheitsinduzierte Signalgebung orchestrieren und Genexpressionsnetzwerke, die Invasivität und andere charakteristische Merkmale hervorrufen ( 71 ).
Zusätzlich zu solchen regulatorischen Mechanismen, die durch die physikalische Tumormikroumgebung ausgestattet sind, kann parakrine Signalgebung, die lösliche Faktoren umfasst, die von den verschiedenen Zelltypen, die feste Tumore bevölkern, in das extrazelluläre Milieu freigesetzt werden, auch zur Induktion mehrerer morphologisch unterschiedlicher invasiver Wachstumsprogramme beitragen ( 72 ), nur eines von denen – als „mesenchymal“ bezeichnet – der oben erwähnte epigenetische Regulationsmechanismus der EMT beteiligt zu sein scheint.
Epigenetische regulatorische Heterogenität
Eine wachsende Wissensbasis erhöht die Wertschätzung für die Bedeutung der intratumoralen Heterogenität bei der Erzeugung der phänotypischen Vielfalt, bei der die geeignetsten Zellen für eine proliferative Expansion und Invasion ihren Brüdern entwachsen und daher für eine maligne Progression ausgewählt werden. Sicherlich ist eine Facette dieser phänotypischen Heterogenität in chronischer oder episodischer genomischer Instabilität und daraus resultierender genetischer Heterogenität in den Zellen begründet, die einen Tumor bevölkern.
Darüber hinaus wird zunehmend deutlich, dass es eine nicht auf Mutationen basierende epigenetische Heterogenität geben kann. Ein herausragendes Beispiel ist das Linker-Histon H1.0, das in Subpopulationen von Krebszellen innerhalb einer Reihe von Tumorarten dynamisch exprimiert und reprimiert wird, mit der daraus folgenden Sequestrierung bzw. Zugänglichkeit von Domänen in Megabasengröße.73 ). Bemerkenswerterweise wurde festgestellt, dass die Population von Krebszellen mit reprimiertem H1.0 stammähnliche Eigenschaften, eine verbesserte tumorinitiierende Fähigkeit und eine Assoziation mit einer schlechten Prognose bei Patienten aufweist.
Ein weiteres Beispiel für epigenetisch regulierte Plastizität wurde in menschlichen oralen Plattenepithelkarzinomen (SCC) beschrieben, bei denen Krebszellen an den invasiven Rändern einen partiellen EMT-Zustand (p-EMT) annehmen, dem die oben genannten mesenchymalen TFs fehlen, aber andere EMT-definierende Gene exprimieren, die vorhanden sind nicht im zentralen Kern der Tumore exprimiert ( 74 ).
Die p-EMT-Zellen stellen offensichtlich keine klonale Kompartimentierung von mutationsveränderten Zellen dar: Kulturen von primären tumorabgeleiteten Krebszellen enthalten dynamische Mischungen von sowohl p-EMT hi- als auch p-EMT lo – Zellen und wenn p-EMT hi/lo – Zellen FACS-gereinigt und kultiviert wurden, kehrten beide zu gemischten Populationen von p-EMT hi und p-EMT lo zurück innerhalb von 4 Tagen. Obwohl parakrine Signale aus dem angrenzenden Stroma als deterministisch für den p-EMT hi – Zustand angesehen werden könnten, spricht das stabile Vorhandensein und die Regeneration der beiden epigenetischen Zustände in der Kultur für einen krebszelleigenen Mechanismus. Bemerkenswerterweise wird diese Schlussfolgerung durch die Analyse von 198 Zelllinien gestützt, die 22 Krebsarten repräsentieren, einschließlich SCC, wobei 12 stabil heterogene epigenetische Zustände (einschließlich der p-EMT in SCC) in den Zelllinienmodellen sowie ihren verwandten Primärtumoren auf verschiedene Weise nachgewiesen wurden (75).
Auch hier konnten die heterogenen phänotypischen Zustände nicht mit nachweisbaren genetischen Unterschieden in Verbindung gebracht werden, und in mehreren Fällen wurde gezeigt, dass FACS-sortierte Zellen eines bestimmten Zustands bei der Kultur dynamisch wieder ins Gleichgewicht kommen, was ein stabiles Gleichgewicht zwischen den heterogenen Zuständen rekapituliert, die in den ursprünglichen Zelllinien beobachtet wurden.
Darüber hinaus erhellen Technologien zur genomweiten Profilerstellung verschiedener Attribute – über die DNA-Sequenz und ihre Mutationsvariation hinaus – einflussreiche Elemente der Annotation und Organisation des Krebszellgenoms, die mit der Patientenprognose und zunehmend mit charakteristischen Fähigkeiten korrelieren ( 76–78 ). Epigenomische Heterogenität wird durch immer leistungsfähigere Technologien zum Profilieren von genomweiter DNA-Methylierung ( 79, 80 ), Histonmodifikation ( 81 ), Chromatinzugänglichkeit ( 82 ) und posttranskriptionaler Modifikation und Translation von RNA ( 83, 84 ) aufgedeckt).
Eine Herausforderung in Bezug auf das hier betrachtete Postulat wird darin bestehen, festzustellen, welche epigenomischen Modifikationen bei bestimmten Krebsarten (i) regulatorische Bedeutung haben und (ii) repräsentativ für eine rein nicht-mutationsbedingte Reprogrammierung sind, im Gegensatz zu einer mutationsgetriebenen und somit durch das Genom erklärbaren Instabilität.
Epigenetische Regulation der Stromazelltypen, die die Tumormikroumgebung bevölkern
Im Allgemeinen wird nicht angenommen, dass die akzessorischen Zellen in der Mikroumgebung des Tumors, die funktionell zum Erwerb charakteristischer Fähigkeiten beitragen, unter genetischer Instabilität und mutationsbedingter Neuprogrammierung leiden, um ihre tumorfördernden Aktivitäten zu verstärken; vielmehr wird gefolgert, dass diese Zellen – krebsassoziierte Fibroblasten, angeborene Immunzellen und Endothelzellen und Perizyten des Tumorgefäßsystems – bei ihrer Rekrutierung durch lösliche und physikalische Faktoren, die die solide Tumormikroumgebung definieren, epigenetisch reprogrammiert werden (2, 85).
Es ist zu erwarten, dass die Multi-Omic-Profiling-Technologien, die derzeit auf Krebszellen angewendet werden, zunehmend zur Untersuchung der akzessorischen (stromalen) Zellen in Tumoren verwendet werden, um aufzuklären, wie normale Zellen beschädigt werden, um die Tumorentwicklung und -progression funktionell zu unterstützen. Beispielsweise eine aktuelle Studie (86) legt nahe, dass eine solche Reprogrammierung neben dem induktiven Austausch von Zytokinen, Chemokinen und Wachstumsfaktoren, die intrazelluläre Signalnetzwerke in all diesen Zelltypen verändern, Modifikationen des Epigenoms beinhalten kann:
Wenn Mausmodelle mit Lungenmetastasen mit einer Kombination aus behandelt wurden ein DNA-Methyltransferase-Inhibitor (5-Azacytidin) und ein Inhibitor der Histonmodifikation (ein HDAC), wurde festgestellt, dass die infiltrierenden myeloischen Zellen von einem unreifen (tumorfördernden) Vorläuferzustand in Zellen übergegangen sind, die reifen interstitiellen (tumorantagonisierenden) Makrophagen ähneln , die im Gegensatz zu ihren Gegenstücken in unbehandelten Tumoren nicht in der Lage waren, die typischen Fähigkeiten zu unterstützen, die für eine effiziente Metastasenkolonisation erforderlich sind ( 86). Es ist vorstellbar, dass Multi-Omic-Profiling und pharmakologische Störungen dazu dienen werden, den neu programmierten epigenetischen Zustand in solchen myeloischen Zellen sowie anderen charakteristischen akzessorischen Zelltypen aufzuklären, die Tumormikroumgebungen bevölkern.
Zusammenfassung
Zusammengenommen unterstützen diese anschaulichen Schnappschüsse die These, dass epigenetische Neuprogrammierung ohne Mutation als echtes ermöglichendes Merkmal akzeptiert werden wird, das dazu dient, den Erwerb charakteristischer Fähigkeiten zu erleichtern ( Abb. 3), die sich von der genomischen DNA-Instabilität und -Mutation unterscheidet. Insbesondere kann erwartet werden, dass sich die nicht mutationsbedingte epigenetische Reprogrammierung als integraler Bestandteil der Ermöglichung der oben diskutierten vorläufigen neuen charakteristischen Fähigkeit der phänotypischen Plastizität erweisen wird, insbesondere als treibende Kraft bei der dynamischen transkriptomischen Heterogenität, die zunehmend gut dokumentiert wird in bösartigen Krebszellen TMEs. Der Fortschritt von Single-Cell-Multi-Omic-Profiling-Technologien soll die jeweiligen Beiträge und das Zusammenspiel zwischen mutationsgetriebener und nicht-mutationsbedingter epigenetischer Regulation bei der Entwicklung von Tumoren während der malignen Progression und Metastasierung beleuchten.
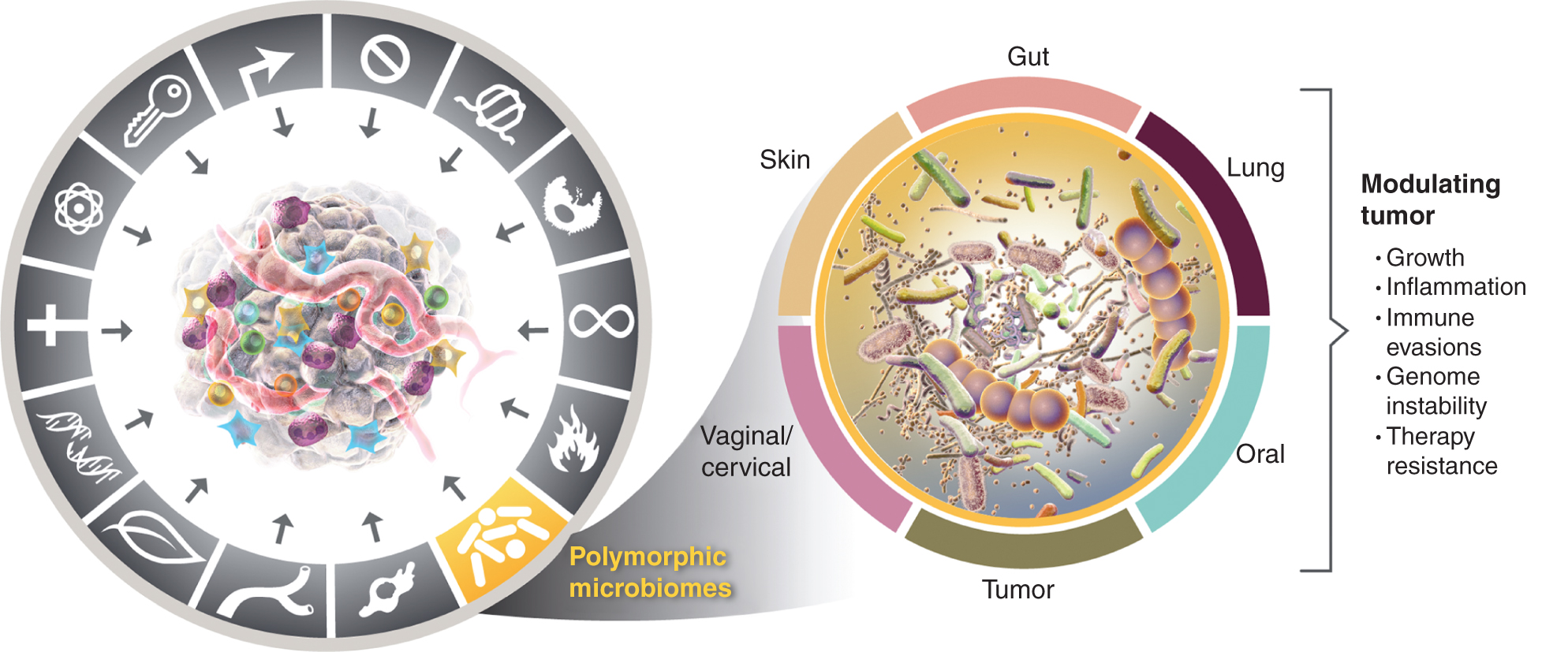
Polymorphe Mikrobiome
Eine weitreichende Grenze in der Biomedizin entfaltet sich durch die Beleuchtung der Vielfalt und Variabilität der Fülle von Mikroorganismen, die zusammen als Mikrobiota bezeichnet werden und die sich symbiotisch mit den Barrieregeweben des Körpers verbinden, die der äußeren Umgebung ausgesetzt sind – insbesondere der Epidermis und der inneren Schleimhaut des Gastrointestinaltrakts sowie der Lunge, der Brust und des Urogenitalsystems.
Es wird zunehmend anerkannt, dass die von ansässigen Bakterien und Pilzen geschaffenen Ökosysteme – die Mikrobiome – tiefgreifende Auswirkungen auf Gesundheit und Krankheit haben (87), eine Erkenntnis, die durch die Fähigkeit vorangetrieben wird, die Populationen mikrobieller Arten mithilfe von Sequenzierungs- und Bioinformatiktechnologien der nächsten Generation zu prüfen. Für Krebs wird der Beweis zunehmend überzeugend, dass polymorphe Variabilität in den Mikrobiomen zwischen Individuen in einer Population tiefgreifende Auswirkungen auf Krebsphänotypen haben kann (88, 89).
Assoziationsstudien in menschlichen und experimentellen Manipulationen in Mausmodellen von Krebs zeigen bestimmte Mikroorganismen, hauptsächlich, aber nicht ausschließlich Bakterien, die entweder schützende oder schädliche Wirkungen auf die Krebsentwicklung, das maligne Fortschreiten und das Ansprechen auf eine Therapie haben können. Dies gilt auch für die globale Komplexität und Zusammensetzung eines Gewebemikrobioms insgesamt. Während das Darmmikrobiom der Pionier dieser neuen Grenze war, haben mehrere Gewebe und Organe Mikrobiome assoziiert, die charakteristische Merkmale in Bezug auf die Populationsdynamik und die Vielfalt mikrobieller Arten und Unterarten aufweisen.
Diese wachsende Wertschätzung der Bedeutung polymorph variabler Mikrobiome für Gesundheit und Krankheit wirft die Frage auf: Ist das Mikrobiom ein eigenständiges ermöglichendes Merkmal, das sich umfassend auswirkt, sowohl positiv als auch negativ, der Erwerb charakteristischer Fähigkeiten für Krebs? Ich denke weiter unten über diese Möglichkeit nach und veranschauliche Beweise für einige der prominenten Gewebemikrobiome, die an Krebsmerkmalen beteiligt sind (Abb. 4), beginnend mit dem prominentesten und offensichtlich wirkungsvollsten Mikrobiom, dem des Darmtrakts.
Abbildung 4
Polymorphe Mikrobiome
Links, während sich die befähigenden Eigenschaften von tumorfördernder Entzündung und genomischer Instabilität und Mutation überschneiden, gibt es zunehmend Grund zu der Schlussfolgerung, dass polymorphe Mikrobiome in einem Individuum im Vergleich zu einem anderen im Dickdarm, in anderen Schleimhäuten und verbundenen Organen oder in Tumoren selbst angesiedelt sind , können viele der charakteristischen Fähigkeiten auf vielfältige Weise beeinflussen – entweder durch Induktion oder Hemmung – und sind daher möglicherweise eine instrumentelle und quasi unabhängige Variable im Rätsel, wie sich Krebs entwickelt, fortschreitet und auf eine Therapie anspricht. Richtig, mehrere Gewebemikrobiome sind an der Modulation von Tumorphänotypen beteiligt. Neben dem weithin untersuchten Darmmikrobiom, anderen charakteristischen Gewebemikrobiomen sowie dem Tumormikrobiom, sind an der Modulation des Erwerbs – sowohl positiv als auch negativ – der dargestellten charakteristischen Fähigkeiten bei bestimmten Tumorarten beteiligt. Die Markenzeichen der Krebsgrafik wurden von Hanahan und Weinberg (2) übernommen.
Vielfältige modulierende Wirkungen des Darmmikrobioms
Es ist seit langem bekannt, dass das Darmmikrobiom für die Funktion des Dickdarms (Kolon) beim Abbau und Import von Nährstoffen in den Körper als Teil der metabolischen Homöostase von grundlegender Bedeutung ist und dass es zu Störungen der mikrobiellen Populationen – Dysbiose – im Dickdarm kommen kann ein Spektrum physiologischer Krankheiten verursachen ( 87). Dazu gehört der Verdacht, dass die Anfälligkeit, Entwicklung und Pathogenese von Dickdarmkrebs durch das Darmmikrobiom beeinflusst wird. In den letzten Jahren haben überzeugende funktionelle Studien mit Stuhltransplantationen von Dickdarmtumor-tragenden Patienten und Mäusen in Empfängermäuse, die für die Entwicklung von Dickdarmkrebs prädisponiert sind, ein Prinzip etabliert: Es gibt sowohl krebsschützende als auch tumorfördernde Mikrobiome, an denen bestimmte Bakterienarten beteiligt sind, die kann das Auftreten und die Pathogenese von Dickdarmtumoren modulieren (90).
Die Mechanismen, durch die Mikrobiota diese modulierenden Rollen übertragen, werden noch aufgeklärt, aber zwei allgemeine Wirkungen sind für tumorfördernde Mikrobiome und in einigen Fällen für spezifische tumorfördernde Bakterienarten zunehmend gut etabliert. Der erste Effekt ist die Mutagenese des Dickdarmepithels als Folge der Produktion von bakteriellen Toxinen und anderen Molekülen, die entweder die DNA direkt schädigen oder die Systeme stören, die die genomische Integrität aufrechterhalten, oder Zellen auf andere Weise stressen, was indirekt die Genauigkeit der DNA-Replikation beeinträchtigt und Reparatur. Ein typisches Beispiel ist E. coli , das den PKS -Locus trägt, der nachweislich das menschliche Genom mutagenisiert und an der Übertragung von Mutationen beteiligt ist, die das Kennzeichen ermöglichen (91).
Außerdem wurde berichtet, dass Bakterien an die Oberfläche von Dickdarmepithelzellen binden und Ligandenmimetika produzieren, die die Epithelproliferation stimulieren, was in neoplastischen Zellen zu der charakteristischen Fähigkeit zur proliferativen Signalübertragung beiträgt (88). Ein weiterer Mechanismus, durch den spezifische Bakterienarten die Tumorentstehung fördern, sind Butyrat-produzierende Bakterien, deren Vorkommen bei Patienten mit kolorektalem Karzinom erhöht ist (92).
Die Produktion des Metaboliten Butyrat hat komplexe physiologische Wirkungen, einschließlich der Induktion alternder Epithel- und Fibroblastenzellen. Ein Mausmodell der Kolonkarzinogenese, das mit Butyrat-produzierenden Bakterien besiedelt war, entwickelte mehr Tumore als Mäuse, denen solche Bakterien fehlten; Der Zusammenhang zwischen Butyrat-induzierter Seneszenz und verstärkter Kolontumorgenese wurde durch die Verwendung eines senolytischen Medikaments gezeigt, das seneszente Zellen abtötet, was das Tumorwachstum beeinträchtigt (92).
Darüber hinaus hat bakteriell produziertes Butyrat pleiotrope und paradoxe Wirkungen auf differenzierte Zellen im Vergleich zu undifferenzierten (Stamm-)Zellen im Dickdarmepithel bei Zuständen, bei denen die Darmbarriere gestört ist (Dysbiose) und die Bakterien invasiv sind, was beispielsweise die Zellenergie beeinflusst und Metabolismus, Histonmodifikation, Zellzyklusprogression und (tumorfördernde) angeborene Immunentzündung, die adaptive Immunantworten immunsupprimiert (93).
In der Tat beinhaltet eine breite Wirkung polymorpher Mikrobiome die Modulation des adaptiven und angeborenen Immunsystems über vielfältige Wege, einschließlich der Produktion von „immunmodulatorischen“ Faktoren durch Bakterien, die Schadenssensoren auf epithelialen oder residenten Immunzellen aktivieren, was zur Expression einer Vielzahl führt Repertoire an Chemokinen und Zytokinen, die die Fülle und Eigenschaften von Immunzellen formen können, die das Dickdarmepithel und das darunter liegende Stroma und die drainierenden Lymphknoten bevölkern.
Darüber hinaus können bestimmte Bakterien sowohl den schützenden Biofilm als auch den Schleim, der das Dickdarmepithel auskleidet, durchbrechen und die epithelialen Zell-Zell-Tight-Junctions stören, die gemeinsam die Integrität der physikalischen Barriere aufrechterhalten, die normalerweise das Darmmikrobiom unterteilt. Beim Eindringen in das Stroma Bakterien können sowohl angeborene als auch adaptive Immunantworten auslösen, indem sie die Sekretion eines Repertoires von Zytokinen und Chemokinen hervorrufen. Eine Manifestation kann die Schaffung tumorfördernder oder tumorantagonisierender Immunmikroumgebungen sein, die folglich vor Tumorentstehung und maligner Progression schützen oder diese erleichtern.
Dementsprechend kann die Modulation der miteinander verflochtenen Parameter von (i) der Auslösung (angeborener) tumorfördernder Entzündungen und (ii) dem Entkommen der (adaptiven) Immunzerstörung durch charakteristische Mikrobiome bei einzelnen Patienten nicht nur mit der Prognose, sondern auch mit dem Ansprechen oder der Resistenz in Verbindung gebracht werden Immuntherapien mit Immuncheckpoint-Inhibitoren und anderen therapeutischen Modalitäten ( Eine Manifestation kann die Schaffung tumorfördernder oder tumorantagonisierender Immunmikroumgebungen sein, die folglich vor Tumorentstehung und maligner Progression schützen oder diese erleichtern.
Dementsprechend kann die Modulation der miteinander verflochtenen Parameter von (i) der Auslösung (angeborener) tumorfördernder Entzündungen und (ii) dem Entkommen der (adaptiven) Immunzerstörung durch charakteristische Mikrobiome bei einzelnen Patienten nicht nur mit der Prognose, sondern auch mit dem Ansprechen oder der Resistenz in Verbindung gebracht werden Immuntherapien mit Immuncheckpoint-Inhibitoren und anderen therapeutischen Modalitäten (Eine Manifestation kann die Schaffung tumorfördernder oder tumorantagonisierender Immunmikroumgebungen sein, die folglich vor Tumorentstehung und maligner Progression schützen oder diese erleichtern).
Dementsprechend kann die Modulation der miteinander verflochtenen Parameter von (i) der Auslösung (angeborener) tumorfördernder Entzündungen und (ii) dem Entkommen der (adaptiven) Immunzerstörung durch charakteristische Mikrobiome bei einzelnen Patienten nicht nur mit der Prognose, sondern auch mit dem Ansprechen oder der Resistenz in Verbindung gebracht werden Immuntherapien mit Immuncheckpoint-Inhibitoren und anderen therapeutischen Modalitäten (89, 94–96 ). Ein vorläufiger Proof-of-Concept stammt aus jüngsten Studien, die die wiederhergestellte Wirksamkeit der Immuntherapie nach Transplantationen fäkaler Mikrobiota von auf die Therapie ansprechenden Patienten in Patienten mit Melanom zeigen, die während einer vorherigen Behandlung mit Immun-Checkpoint-Blockade fortgeschritten waren ( 97, 98 ).
Die molekularen Mechanismen, durch die bestimmte und variable Bestandteile des Darmmikrobioms die Aktivität des adaptiven Immunsystems systemisch modulieren, sind ein anhaltendes Rätsel, indem sie entweder die durch die Blockade der Immuncheckpoints hervorgerufenen antitumoralen Immunantworten verstärken oder vielmehr eine systemische oder lokale (intratumorale) Immunsuppression hervorrufen. Eine kürzlich durchgeführte Studie hat Licht ins Dunkel gebracht: Bestimmte Stämme von Enterococcus (und anderen Bakterien) exprimieren eine Peptidoglycan-Hydrolyase namens SagA, die Mucopeptide aus der Bakterienwand freisetzt, die dann systemisch zirkulieren und den NOD2-Musterrezeptor aktivieren können, was wiederum T- Zellreaktionen und die Wirksamkeit der Checkpoint-Immuntherapie (99).
Andere immunregulatorische Moleküle, die von spezifischen bakteriellen Unterarten produziert werden, werden identifiziert und funktionell bewertet, einschließlich von Bakterien produziertem Inosin, einem geschwindigkeitsbestimmenden Metaboliten für die T-Zell-Aktivität (100). Diese und andere Beispiele beginnen, die molekularen Mechanismen aufzuzeichnen, durch die polymorphe Mikrobiome indirekt und systemisch die Tumorimmunbiologie modulieren, und zwar über und über Immunantworten hinaus, die auf direkte physikalische Wechselwirkungen von Bakterien mit dem Immunsystem folgen (101, 102 ).
Abgesehen von den kausalen Verbindungen zu Dickdarmkrebs und Melanom ist die nachweisbare Fähigkeit des Darmmikrobioms, die Expression von immunmodulatorischen Chemokinen und Zytokinen hervorzurufen, die in den systemischen Kreislauf gelangen, offensichtlich auch in der Lage, die Krebspathogenese und das Ansprechen auf Therapien in anderen Organen des Körpers zu beeinflussen (94, 95 ).
Ein aufschlussreiches Beispiel betrifft die Entwicklung von Cholangiokarzinomen in der Leber: Darmdysbiose ermöglicht den Eintritt und Transport von Bakterien und bakteriellen Produkten durch die Pfortader zur Leber, wo auf Hepatozyten exprimiertes TLR4 ausgelöst wird, um die Expression des Chemokins CXCL1 zu induzieren, das CXCR2 rekrutiert -exprimierende granulozytäre myeloische Zellen (gMDSC), die dazu dienen, natürliche Killerzellen zu unterdrücken, um der Immunzerstörung zu entgehen (103) und vermitteln wahrscheinlich andere charakteristische Fähigkeiten (85). Als solches ist das Darmmikrobiom eindeutig als ein ermöglichendes Merkmal impliziert, das mehrere Krebsarten alternativ erleichtern oder davor schützen kann.
Jenseits des Darms: Implizieren ausgeprägter Mikrobiome in anderen Barrieregeweben
Nahezu alle Gewebe und Organe, die direkt oder indirekt der Außenumgebung ausgesetzt sind, sind auch Aufbewahrungsorte für kommensale Mikroorganismen ( 104 ). Im Gegensatz zum Darm, wo die symbiotische Rolle des Mikrobioms im Stoffwechsel gut anerkannt ist, ist die normale und pathogene Rolle der ansässigen Mikrobiota an diesen verschiedenen Orten noch im Entstehen.
Es gibt offensichtlich organ-/gewebespezifische Unterschiede in der Konstitution der assoziierten Mikrobiome bei Homöostase, Alterung und Krebs, mit sowohl überlappenden als auch charakteristischen Arten und Häufigkeiten zu denen des Dickdarms ( 104, 105). Darüber hinaus liefern Assoziationsstudien zunehmend Hinweise darauf, dass lokale tumorantagonisierende/protektive versus tumorfördernde Gewebemikrobiome, ähnlich wie das Darmmikrobiom, die Anfälligkeit und Pathogenese für menschliche Krebserkrankungen modulieren können, die in ihren assoziierten Organen entstehen ( 106–109 ).
Einfluss der intratumoralen Mikrobiota?
Schließlich haben Pathologen schon lange erkannt, dass Bakterien in soliden Tumoren nachgewiesen werden können, eine Beobachtung, die jetzt durch ausgefeilte Profiling-Technologien untermauert wurde. Beispielsweise war in einer Untersuchung von 1.526 Tumoren, die sieben menschliche Krebsarten umfassten (Knochen, Gehirn, Brust, Lunge, Melanom, Eierstöcke und Bauchspeicheldrüse), jede Art durch ein charakteristisches Mikrobiom gekennzeichnet, das größtenteils in Krebszellen und Immunzellen lokalisiert war. Iinnerhalb jedes Tumortyps konnten Variationen im Tumormikrobiom nachgewiesen und daraus geschlossen werden, dass sie mit klinisch-pathologischen Merkmalen assoziiert sind (110).
Mikrobiota wurden in ähnlicher Weise in gentechnisch veränderten de novo nachgewiesenMausmodellen von Lungen- und Bauchspeicheldrüsenkrebs, und ihr Fehlen in keimfreien Mäusen und/oder ihre Aufhebung mit Antibiotika kann die Tumorentstehung nachweislich beeinträchtigen, was das Tumormikrobiom funktionell als Wegbereiter für tumorfördernde Entzündungen und maligne Progression impliziert (111, 112).
Assoziationsstudien beim humanen duktalen Adenokarzinom des Pankreas und Funktionstests über Stuhltransplantationen in tumortragende Mäuse haben gezeigt, dass Variationen im Tumormikrobiom – und dem damit verbundenen Darmmikrobiom – die Phänotypen des Immunsystems und das Überleben modulieren (113). Eine wichtige Herausforderung für die Zukunft wird darin bestehen, diese Implikationen auf andere Tumorarten auszudehnen und die möglicherweise trennbaren Beiträge der Konstitution und Variation im Tumormikrobiom von denen des Darmmikrobioms (und des lokalen Ursprungsgewebes) abzugrenzen, möglicherweise durch Identifizierung spezifischer mikrobielle Arten, die an dem einen oder anderen Ort funktionell einflussreich sind.
Zusammenfassung
Zu den faszinierenden Fragen für die Zukunft gehört, ob Mikrobiota, die in verschiedenen Geweben ansässig sind oder beginnende Neoplasien bevölkern, die Fähigkeit haben, zum Erwerb anderer charakteristischer Fähigkeiten jenseits von Immunmodulation und Genommutation beizutragen oder diese zu stören und dadurch die Tumorentwicklung und -progression zu beeinflussen. Es gibt Hinweise darauf, dass bestimmte Bakterienspezies das Markenzeichen der proliferativen Signalübertragung direkt stimulieren können, beispielsweise im Dickdarmepithel (88), und die Wachstumsunterdrückung modulieren können, indem sie die Tumorsuppressoraktivität in verschiedenen Kompartimenten des Darms verändern (114), während direkte Wirkungen auf andere charakteristische Fähigkeiten, wie das Vermeiden von Zelltod, das Auslösen von Angiogenese und das Stimulieren von Invasion und Metastasierung, unklar bleiben, ebenso wie die Verallgemeinerbarkeit dieser Beobachtungen auf mehrere Formen von menschlichem Krebs.
Unabhängig davon gibt es immer überzeugendere Argumente dafür, dass die polymorphe Variation in den Mikrobiomen des Darms und anderer Organe ein charakteristisches Aktivierungsmerkmal für den Erwerb charakteristischer Fähigkeiten darstellt ( Abb. 4 ), auch wenn sie sich mit denen der Genominstabilität und -mutation überschneidet und diese ergänzt , und tumorfördernde Entzündungen.
Seneszente Zellen
Die zelluläre Seneszenz ist eine typischerweise irreversible Form des proliferativen Stillstands, die sich wahrscheinlich als Schutzmechanismus zur Aufrechterhaltung der Gewebehomöostase entwickelt hat, angeblich als komplementärer Mechanismus zum programmierten Zelltod, der dazu dient, erkrankte, funktionsgestörte oder anderweitig unnötige Zellen zu inaktivieren und zu gegebener Zeit zu entfernen. Zusätzlich zum Abschalten des Zellteilungszyklus ruft das Seneszenzprogramm Veränderungen in der Zellmorphologie und im Stoffwechsel hervor und, am tiefsten, die Aktivierung eines Seneszenz-assoziierten sekretorischen Phänotyps (SASP), der die Freisetzung einer Fülle von bioaktiven Proteinen, einschließlich Chemokinen, beinhaltet.
Zytokine und Proteasen, deren Identität vom Zell- und Gewebetyp abhängt, aus dem eine seneszente Zelle hervorgeht ( 115–117). Seneszenz kann in Zellen durch eine Vielzahl von Bedingungen induziert werden, einschließlich Mikroumgebungsstress wie Nährstoffmangel und DNA-Schäden sowie Schäden an Organellen und zellulärer Infrastruktur und Ungleichgewichte in zellulären Signalnetzwerken ( 115, 117 ), die alle aufgetreten sind im Zusammenhang mit der beobachteten Zunahme der Häufigkeit seneszenter Zellen in verschiedenen Organen während des Alterns ( 118, 119 ).
Zelluläre Seneszenz wird seit langem als Schutzmechanismus gegen Neoplasien angesehen, wodurch Krebszellen dazu gebracht werden, sich einer Seneszenz zu unterziehen ( 120 ). Die meisten der oben genannten Initiatoren des Seneszenzprogramms sind mit Malignität assoziiert, insbesondere DNA-Schäden als Folge einer aberranten Hyperproliferation, sogenannter onkogeninduzierter Seneszenz aufgrund einer hyperaktivierten Signalübertragung und einer therapieinduzierten Seneszenz als Folge einer zellulären und genomischen Schädigung verursacht durch Chemotherapie und Strahlentherapie.
Tatsächlich gibt es gut etablierte Beispiele für die schützenden Vorteile der Seneszenz bei der Begrenzung der malignen Progression (118, 119). Im Gegenteil, eine zunehmende Zahl von Beweisen zeigt jedoch genau das Gegenteil: In bestimmten Kontexten stimulieren seneszente Zellen auf unterschiedliche Weise die Tumorentwicklung und maligne Progression (119, 121 ).
In einer aufschlussreichen Fallstudie wurden seneszente Zellen in alternden Mäusen pharmakologisch abgetragen, insbesondere seneszente Zellen depletiert, die charakteristischerweise den Zellzyklus-Inhibitor p16 – INK4a exprimieren : Zusätzlich zur Verzögerung mehrerer altersbedingter Symptome führte dies zu einer Depletion seneszenter Zellen in alternden Mäusen bei reduzierten Inzidenzen von spontaner Tumorentstehung und krebsassoziiertem Tod (122).
Es wird angenommen, dass der Hauptmechanismus, durch den seneszente Zellen Tumorphänotypen fördern, das SASP ist, das nachweislich in der Lage ist, auf parakrine Weise Signalmoleküle (und Proteasen, die das aktivieren und/oder deaktivieren), um typische Fähigkeiten zu vermitteln. Somit wurde in verschiedenen experimentellen Systemen gezeigt, dass seneszente Krebszellen auf verschiedene Weise zur proliferativen Signalgebung beitragen, Apoptose vermeiden, Angiogenese induzieren, Invasion und Metastasierung stimulieren und Tumorimmunität unterdrücken (116, 118, 120, 121).
Noch eine weitere Facette der Auswirkungen von seneszenten Krebszellen auf Krebsphänotypen beinhaltet vorübergehende, reversible seneszente Zellzustände, wodurch seneszente Krebszellen aus ihrem SASP-exprimierenden, nicht proliferativen Zustand entkommen und die Zellproliferation und Manifestation der damit verbundenen Fähigkeiten eines vollständig lebensfähigen Onkogens wieder aufnehmen können Zellen (44).
Eine solche vorübergehende Seneszenz ist am besten in Fällen von Therapieresistenz dokumentiert (44), die eine Form der Ruhe darstellt, die das therapeutische Targeting von proliferierenden Krebszellen umgeht, sich aber in anderen Stadien der Tumorentwicklung, der malignen Progression und in anderen Stadien der Tumorentwicklung als breiter wirksam erweisen könnte Metastasierung.
Darüber hinaus sind die Kennzeichen-fördernden Fähigkeiten seneszenter Zellen nicht auf seneszente Krebszellen beschränkt. Es wurde gezeigt, dass krebsassoziierte Fibroblasten (CAF) in Tumoren seneszieren, wodurch seneszente CAFs entstehen, die nachweislich tumorfördernd sind, indem sie charakteristische Fähigkeiten an Krebszellen im TME weitergeben (115, 116, 121).
Darüber hinaus sind seneszente Fibroblasten in normalen Geweben, die teilweise durch natürliches Altern oder Umwelteinflüsse entstanden sind, in ähnlicher Weise an der Umgestaltung von Gewebemikroumgebungen über ihre SASP beteiligt, um eine parakrine Unterstützung für lokale Invasionen (sogenannte „Feldeffekte“) und Fernmetastasen bereitzustellen (116) von Neoplasien, die sich in der Nähe entwickeln.
Darüber hinaus wurde gezeigt, dass seneszente Fibroblasten in alternder Haut – über ihre SASP – angeborene Immunzellen rekrutieren, die sowohl immunsuppressiv für adaptive antitumorale Immunantworten sind, die durch CD8-T-Zellen verankert sind, als auch das Wachstum von Hauttumoren stimulieren (123), wobei letztere Wirkung möglicherweise besteht was parakrine Beiträge solcher angeborener Immunzellen (myeloische Zellen, Neutrophile und Makrophagen) zu anderen charakteristischen Fähigkeiten widerspiegelt.
Obwohl weniger gut etabliert, scheint es wahrscheinlich, dass andere reichlich vorhandene Stromazellen, die bestimmte Tumormikroumgebungen bevölkern, sich als Seneszenz erweisen und dadurch Krebsmerkmale und daraus resultierende Tumorphänotypen modulieren werden. Zum Beispiel können therapieinduzierte seneszente Tumorendothelzellen die Proliferation, Invasion und Metastasierung in Brustkrebsmodellen verstärken (124, 125).
Sicherlich rechtfertigen solche Hinweise eine Untersuchung bei anderen Tumortypen, um die allgemeine Seneszenz von Fibroblasten, Endothelzellen und anderen Stromazellen als treibende Kraft in der Tumorentwicklung zu bewerten. Ebenfalls derzeit ungeklärt sind die regulatorischen Mechanismen und funktionellen Determinanten, durch die ein bestimmter seneszierender Zelltyp in einer bestimmten TME eine tumorfördernde gegenüber einer tumorantagonisierenden SASP hervorruft, die anscheinend alternativ in demselben seneszierenden Zelltyp induziert werden kann, möglicherweise durch verschiedene Initiatoren wenn sie in charakteristische physiologische und neoplastische Mikroumgebungen eingetaucht sind.
Zusammenfassung
Das Konzept, dass Tumore aus genetisch transformierten Krebszellen bestehen, die mit rekrutierten und epigenetisch/phänotypisch korrumpierten akzessorischen (Stroma-)Zellen interagieren und von ihnen profitieren, ist als maßgeblich für die Pathogenese von Krebs etabliert. Die oben diskutierten und in den hier (und anderswo) zitierten Übersichten und Berichten beschriebenen Überlegungen sprechen überzeugend dafür, dass seneszente Zellen (egal welchen zellulären Ursprungs) für die Aufnahme in die Liste der funktionell signifikanten Zellen in der Tumormikroumgebung in Betracht gezogen werden sollten (Abb. 5). Daher sollten seneszente Zellen bei der Suche nach tiefgreifendem Wissen über Krebsmechanismen berücksichtigt werden. Darüber hinaus motiviert die Erkenntnis ihrer Bedeutung das untergeordnete Ziel, tumorfördernde seneszente Zellen aller Konstitutionen therapeutisch anzugreifen, sei es durch pharmakologische oder immunologische Ablation oder durch Reprogrammierung der SASP in tumorantagonisierende Varianten (115, 121, 126).
Abbildung 5

Seneszente Zellen
Heterogene Krebszellsubtypen sowie Stromazelltypen und -subtypen sind funktionell in die Manifestationen von Tumoren als illegale Organe integriert. Hinweise deuten zunehmend darauf hin, dass seneszente Zellderivate vieler dieser zellulären Bestandteile der TME und ihre variablen SASPs an der Modulation von Markenzeichen-Fähigkeiten und daraus resultierenden Tumorphänotypen beteiligt sind. Die Markenzeichen der Krebsgrafik wurden von Hanahan und Weinberg (2) übernommen.
Abschließende Bemerkungen
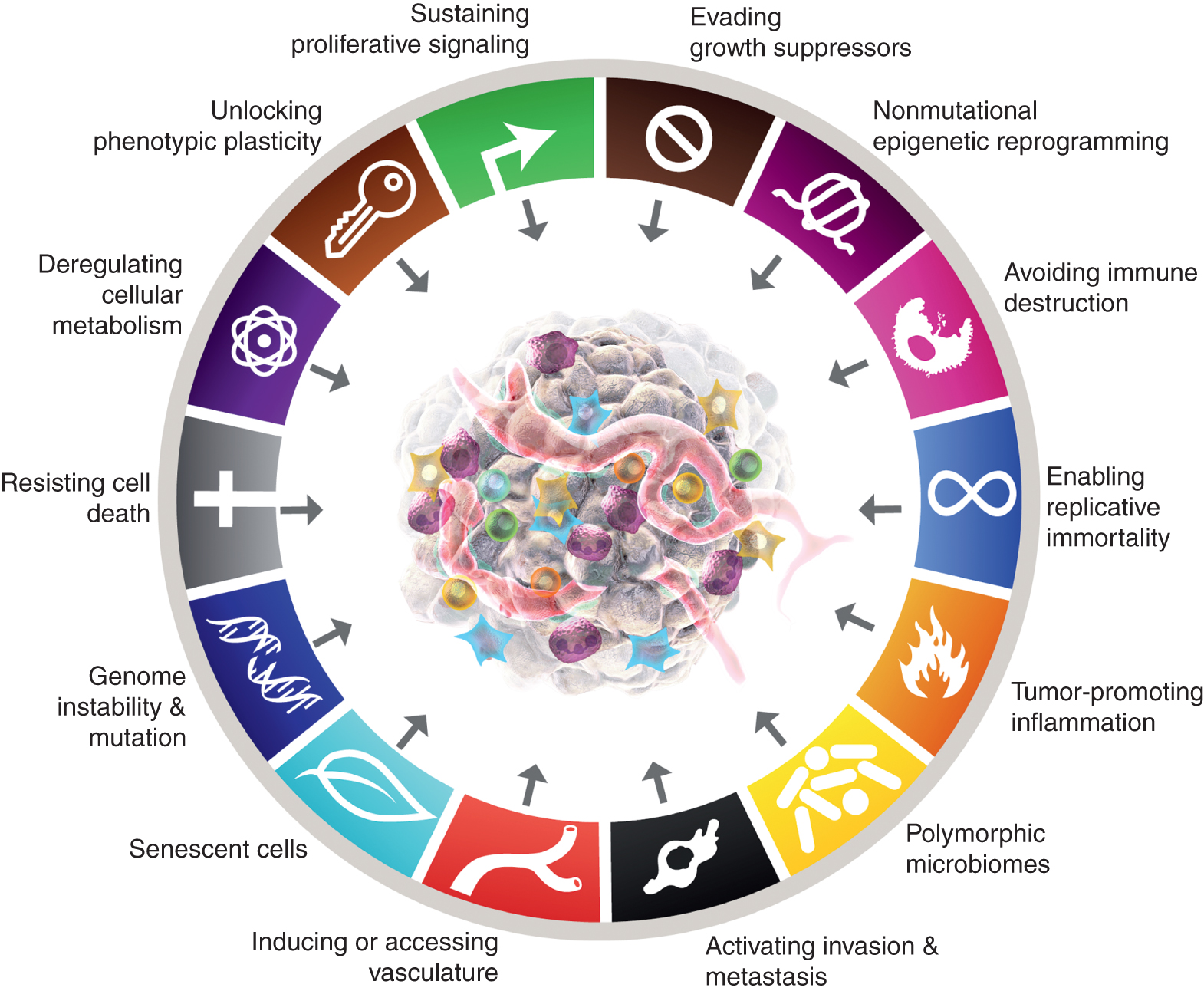
Während sich die acht Markenzeichen von Krebs und ihre beiden unterstützenden Merkmale als dauerhafter heuristischer Wert bei der Konzeptualisierung von Krebs erwiesen haben, deuten die oben dargelegten Überlegungen darauf hin, dass es neue Facetten einer gewissen Allgemeingültigkeit geben könnte und daher von Bedeutung für ein vollständigeres Verständnis der Komplexität, Mechanismen, und Manifestationen der Krankheit. Durch die Anwendung der Metrik der erkennbaren, wenn nicht vollständigen Unabhängigkeit von den 10 Kernattributen ist es vertretbar, dass diese vier Parameter – nach weiterer Validierung und Verallgemeinerung über die vorgestellten Fallstudien hinaus – durchaus in die Kennzeichen des Krebsschemas integriert werden können (Abb. 6).
Daher könnte die zelluläre Plastizität der Liste der herausragenden Fähigkeiten hinzugefügt werden. Während der achte Kern und diese Nouveau-Fähigkeit jeweils durch ihre Definition als Kennzeichen konzeptionell unterscheidbar sind, sind Aspekte ihrer Regulation bei einigen und vielleicht vielen Krebsarten zumindest teilweise miteinander verbunden. Beispielsweise werden mehrere Kennzeichen bei einigen Tumortypen durch kanonische onkogene Treiber koordiniert moduliert, einschließlich
- (I) KRAS ( https://cancer.sanger.ac.uk/cosmic/census-page/KRAS ),
- (II) MYC ( https://cancer.sanger.ac.uk/cosmic/census-page/MYC ),
- (III) NOTCH ( https://cancer.sanger.ac.uk/cosmic/census-page/NOTCH1 ; Ref. 127) und
- (IV) TP53 ( https://cancer.sanger.ac.uk/cosmic/census-page/TP53 )
Abbildung 6
Kennzeichen des Krebs
Dargestellt sind die kanonischen und voraussichtlichen Neuzugänge zu den „Markenzeichen des Krebses“. Diese Abhandlung wirft die Möglichkeit auf, mit dem Ziel, Debatten, Diskussionen und experimentelle Ausarbeitungen anzuregen, dass einige oder alle der vier neuen Parameter als generisch für mehrere Formen von menschlichem Krebs anerkannt werden und daher geeignet sind, sie in die Kernkonzeption des zu integrieren Kennzeichen von Krebs. Die Markenzeichen der Krebsgrafik wurden von Hanahan und Weinberg (2) übernommen.
Zusätzlich zum Hinzufügen von zellulärer Plastizität zum Kader können epigenetische Reprogrammierung ohne Mutation und polymorphe Variationen in Organ-/Gewebemikrobiomen als mechanistische Determinanten – ermöglichende Eigenschaften – integriert werden, durch die charakteristische Fähigkeiten erworben werden, zusammen mit tumorfördernden Entzündungen (selbst teilweise miteinander verbunden zum Mikrobiom), über die Mutationen und andere Aberrationen hinaus, die die oben erwähnten onkogenen Treiber manifestieren.
Schließlich können seneszente Zellen unterschiedlichen Ursprungs – einschließlich Krebszellen und verschiedener Stromazellen – die funktionell zur Entwicklung und bösartigen Progression von Krebs beitragen, wenn auch in deutlich unterschiedlicher Weise zu denen ihrer nicht seneszenten Brüder, als generische Komponenten der TME aufgenommen werden. Zusammenfassend ist vorgesehen, dass das Aufstellen dieser vorläufigen „Versuchsballons“ Debatten, Diskussionen und weitere experimentelle Untersuchungen in der Krebsforschungsgemeinschaft über die definierenden konzeptionellen Parameter der Krebsbiologie, -genetik und -pathogenese anregen wird.
[toggle title=“Verweise“ state=“close“]
- Hanahan D , Weinberg RA . The hallmarks of cancer. Cell 2000;100:57–70.
- Hanahan D , Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Yuan S , Norgard RJ , Stanger BZ . Cellular plasticity in cancer. Cancer Discov 2019;9:837–51.
- Barker N , Ridgway RA , van Es JH , van de Wetering M , Begthel H , van den Born M et al . Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608–11.
- Perekatt AO , Shah PP , Cheung S , Jariwala N , Wu A , Gandhi V et al . SMAD4 suppresses WNT-driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res 2018;78:4878–90.
- Shih IM , Wang TL , Traverso G , Romans K , Hamilton SR , Ben-Sasson S et al . Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A 2001;98:2640–5.
- Ordóñez-Morán P , Dafflon C , Imajo M , Nishida E , Huelsken J . HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 2015;28:815–29.
- Tan SH , Barker N . Stemming colorectal cancer growth and metastasis: HOXA5 forces cancer stem cells to differentiate. Cancer Cell 2015;28:683–5.
- Köhler C , Nittner D , Rambow F , Radaelli E , Stanchi F , Vandamme N et al . Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 2017;21:679–93.
- Shah M , Bhoumik A , Goel V , Dewing A , Breitwieser W , Kluger H et al . A role for ATF2 in regulating MITF and melanoma development. PLoS Genet 2010;6:e1001258.
- Claps G , Cheli Y , Zhang T , Scortegagna M , Lau E , Kim H et al . A transcriptionally inactive ATF2 variant drives melanomagenesis. Cell Rep 2016;15:1884–92.
- Saghafinia S , Homicsko K , Di Domenico A , Wullschleger S , Perren A , Marinoni I et al . Cancer cells retrace a stepwise differentiation program during malignant progression. Cancer Discov 2021;11:2638–57.
- Yu X-X , Qiu W-L , Yang L , Zhang Y , He M-Y , Li L-C et al . Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J 2019;38:e100164.
- de Thé H . Differentiation therapy revisited. Nat Rev Cancer 2018;18:117–27.
- He LZ , Merghoub T , Pandolfi PP . In vivo analysis of the molecular pathogenesis of acute promyelocytic leukemia in the mouse and its therapeutic implications. Oncogene 1999;18:5278–92.
- Warrell RP , de Thé H , Wang ZY , Degos L . Acute promyelocytic leukemia. N Engl J Med 1993;329:177–89.
- Bots M , Verbrugge I , Martin BP , Salmon JM , Ghisi M , Baker A et al . Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014;123:1341–52.
- Ferrara FF , Fazi F , Bianchini A , Padula F , Gelmetti V , Minucci S et al . Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res 2001;61:2–7.
- Kaufman CK , Mosimann C , Fan ZP , Yang S , Thomas AJ , Ablain J et al . A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016;351:aad2197.
- Morris JP , Yashinskie JJ , Koche R , Chandwani R , Tian S , Chen C-C et al . α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019;573:595–9.
- Saha SK , Parachoniak CA , Ghanta KS , Fitamant J , Ross KN , Najem MS et al . Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110–4.
- Dang L , Su S-SM . Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem 2017;86:305–31.
- Waitkus MS , Diplas BH , Yan H . Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018;34:186–95.
- Phan TG , Croucher PI . The dormant cancer cell life cycle. Nat Rev Cancer 2020;20:398–411.
- Jiang M , Azevedo-Pouly AC , Deering TG , Hoang CQ , DiRenzo D , Hess DA et al . MIST1 and PTF1 collaborate in feed-forward regulatory loops that maintain the pancreatic acinar phenotype in adult mice. Mol Cell Biol 2016;36:2945–55.
- Krah NM , Narayanan SM , Yugawa DE , Straley JA , Wright CVE , MacDonald RJ et al . Prevention and reversion of pancreatic tumorigenesis through a differentiation-based mechanism. Dev Cell 2019;50:744–54.
- Krah NM , De La O J-P , Swift GH , Hoang CQ , Willet SG , Chen Pan F et al . The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015;4:e07125.
- Shi G , DiRenzo D , Qu C , Barney D , Miley D , Konieczny SF . Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 2013;32:1950–8.
- Kopp JL , von Figura G , Mayes E , Liu F-F , Dubois CL , Morris JP et al . Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50.
- Julian LM , McDonald AC , Stanford WL . Direct reprogramming with SOX factors: masters of cell fate. Curr Opin Genet Dev 2017;46:24–36.
- Grimm D , Bauer J , Wise P , Krüger M , Simonsen U , Wehland M et al . The role of SOX family members in solid tumours and metastasis. Semin Cancer Biol 2020;67:122–53.
- Mu P , Zhang Z , Benelli M , Karthaus WR , Hoover E , Chen C-C et al . SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84–8.
- Von Hoff DD , LoRusso PM , Rudin CM , Reddy JC , Yauch RL , Tibes R et al . Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72.
- Biehs B , Dijkgraaf GJP , Piskol R , Alicke B , Boumahdi S , Peale F et al . A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018;562:429–33.
- Boumahdi S , de Sauvage FJ . The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19:39–56.
- Groves SM , Ireland A , Liu Q , Simmons AJ , Lau K , Iams WT et al . Cancer Hallmarks Define a Continuum of Plastic Cell States between Small Cell Lung Cancer Archetypes [Internet]. Systems Biology; 2021 Jan. Available from: http://biorxiv.org/lookup/doi/10.1101/2021.01.22.427865.
- LaFave LM , Kartha VK , Ma S , Meli K , Del Priore I , Lareau C et al . Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 2020;38:212–28.
- Marjanovic ND , Hofree M , Chan JE , Canner D , Wu K , Trakala M et al . Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 2020;38:229–46.
- Drapkin BJ , Minna JD . Studying lineage plasticity one cell at a time. Cancer Cell 2020;38:150–2.
- Inoue Y , Nikolic A , Farnsworth D , Liu A , Ladanyi M , Somwar R et al . Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer [Internet]. Cancer Biology; 2020 Nov. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.11.12.368522.
- Dravis C , Chung C-Y , Lytle NK , Herrera-Valdez J , Luna G , Trejo CL et al . Epigenetic and transcriptomic profiling of mammary gland development and tumor models disclose regulators of cell state plasticity. Cancer Cell 2018;34:466–82.
- Malta TM , Sokolov A , Gentles AJ , Burzykowski T , Poisson L , Weinstein JN et al . Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–54.
- Miao Z-F , Lewis MA , Cho CJ , Adkins-Threats M , Park D , Brown JW et al . A dedicated evolutionarily conserved molecular network licenses differentiated cells to return to the cell cycle. Dev Cell 2020;55:178–94.
- De Blander H , Morel A-P , Senaratne AP , Ouzounova M , Puisieux A . Cellular plasticity: a route to senescence exit and tumorigenesis. Cancers 2021;13:4561.
- Merrell AJ , Stanger BZ . Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 2016;17:413–25.
- Baylin SB , Jones PA . Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 2016;8:a019505.
- Flavahan WA , Gaskell E , Bernstein BE . Epigenetic plasticity and the hallmarks of cancer. Science 2017;357:eaal2380.
- Jones PA , Issa J-PJ , Baylin S . Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630–41.
- Huang S . Tumor progression: Chance and necessity in Darwinian and Lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol 2012;110:69–86.
- Darwiche N . Epigenetic mechanisms and the hallmarks of cancer: an intimate affair. Am J Cancer Res 2020;10:1954–78.
- Feng Y , Liu X , Pauklin S . 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell 2021;12:440–54.
- Nam AS , Chaligne R , Landau DA . Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet 2021;22:3–18.
- Bitman-Lotan E , Orian A . Nuclear organization and regulation of the differentiated state. Cell Mol Life Sci CMLS 2021;78:3141–58.
- Goldberg AD , Allis CD , Bernstein E . Epigenetics: a landscape takes shape. Cell 2007;128:635–8.
- Zeng Y , Chen T . DNA methylation reprogramming during mammalian development. Genes 2019;10:257.
- Hegde AN , Smith SG . Recent developments in transcriptional and translational regulation underlying long-term synaptic plasticity and memory. Learn Mem 2019;26:307–17.
- Kim S , Kaang B-K . Epigenetic regulation and chromatin remodeling in learning and memory. Exp Mol Med 2017;49:e281.
- Thienpont B , Van Dyck L , Lambrechts D . Tumors smother their epigenome. Mol Cell Oncol 2016;3:e1240549.
- Gameiro PA , Struhl K . Nutrient deprivation elicits a transcriptional and translational inflammatory response coupled to decreased protein synthesis. Cell Rep 2018;24:1415–24.
- Lin GL , Monje M . Understanding the deadly silence of posterior fossa A ependymoma. Mol Cell 2020;78:999–1001.
- Michealraj KA , Kumar SA , Kim LJY , Cavalli FMG , Przelicki D , Wojcik JB et al . Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell 2020;181:1329–45.
- Bakir B , Chiarella AM , Pitarresi JR , Rustgi AK . EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol 2020;30:764–76.
- Gupta PB , Pastushenko I , Skibinski A , Blanpain C , Kuperwasser C . Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 2019;24:65–78.
- Lambert AW , Weinberg RA . Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer 2021;21:325–38.
- Lindner P , Paul S , Eckstein M , Hampel C , Muenzner JK , Erlenbach-Wuensch K et al . EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis 2020;11:147.
- Javaid S , Zhang J , Anderssen E , Black JC , Wittner BS , Tajima K et al . Dynamic chromatin modification sustains epithelial-mesenchymal transition following inducible expression of Snail-1. Cell Rep 2013;5:1679–89.
- Serrano-Gomez SJ , Maziveyi M , Alahari SK . Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 2016;15:18.
- Skrypek N , Goossens S , De Smedt E , Vandamme N , Berx G . Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet TIG 2017;33:943–59.
- Li L , Hanahan D . Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 2013;153:86–100.
- Li L , Zeng Q , Bhutkar A , Galván JA , Karamitopoulou E , Noordermeer D et al . GKAP acts as a genetic modulator of NMDAR signaling to govern invasive tumor growth. Cancer Cell 2018;33:736–51.
- Mohammadi H , Sahai E . Mechanisms and impact of altered tumour mechanics. Nat Cell Biol 2018;20:766–74.
- Odenthal J , Takes R , Friedl P . Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis 2016;37:1117–28.
- Torres CM , Biran A , Burney MJ , Patel H , Henser-Brownhill T , Cohen A-HS et al . The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 2016;353:aaf1644.
- Puram SV , Tirosh I , Parikh AS , Patel AP , Yizhak K , Gillespie S et al . Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171:1611–24.
- Kinker GS , Greenwald AC , Tal R , Orlova Z , Cuoco MS , McFarland JM et al . Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 2020;52:1208–18.
- Murtha M , Esteller M . Extraordinary cancer epigenomics: thinking outside the classical coding and promoter box. Trends Cancer 2016;2:572–84.
- Nebbioso A , Tambaro FP , Dell’Aversana C , Altucci L . Cancer epigenetics: moving forward. PLoS Genet 2018;14:e1007362.
- Tavernari D , Battistello E , Dheilly E , Petruzzella AS , Mina M , Sordet-Dessimoz J et al . Non-genetic evolution drives lung adenocarcinoma spatial heterogeneity and progression. Cancer Discov 2021;11:1490–507.
- Heyn H , Vidal E , Ferreira HJ , Vizoso M , Sayols S , Gomez A et al . Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 2016;17:11.
- Saghafinia S , Mina M , Riggi N , Hanahan D , Ciriello G . Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 2018;25:1066–80.
- Audia JE , Campbell RM . Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8:a019521.
- Corces MR , Granja JM , Shams S , Louie BH , Seoane JA , Zhou W et al . The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898.
- Esteve-Puig R , Bueno-Costa A , Esteller M . Writers, readers and erasers of RNA modifications in cancer. Cancer Lett 2020;474:127–37.
- Janin M , Coll-SanMartin L , Esteller M . Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer 2020;19:70.
- Hanahan D , Coussens LM . Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–22.
- Lu Z , Zou J , Li S , Topper MJ , Tao Y , Zhang H et al . Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020;579:284–90.
- Thomas S , Izard J , Walsh E , Batich K , Chongsathidkiet P , Clarke G et al . The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res 2017;77:1783–812.
- Dzutsev A , Badger JH , Perez-Chanona E , Roy S , Salcedo R , Smith CK et al . Microbes and cancer. Annu Rev Immunol 2017;35:199–228.
- Helmink BA , Khan MAW , Hermann A , Gopalakrishnan V , Wargo JA . The microbiome, cancer, and cancer therapy. Nat Med 2019;25:377–88.
- Sears CL , Garrett WS . Microbes, microbiota, and colon cancer. Cell Host Microbe 2014;15:317–28.
- Pleguezuelos-Manzano C , Puschhof J , Rosendahl Huber A , van Hoeck A , Wood HM , Nomburg J et al . Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020;580:269–73.
- Okumura S , Konishi Y , Narukawa M , Sugiura Y , Yoshimoto S , Arai Y et al . Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat Commun 2021;12:5674.
- Salvi PS , Cowles RA . Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in homeostasis and disease. Cells 2021;10:1775.
- Fessler J , Matson V , Gajewski TF . Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 2019;7:108.
- Gopalakrishnan V , Helmink BA , Spencer CN , Reuben A , Wargo JA . The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 2018;33:570–80.
- Zitvogel L , Ma Y , Raoult D , Kroemer G , Gajewski TF . The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–70.
- Baruch EN , Youngster I , Ben-Betzalel G , Ortenberg R , Lahat A , Katz L et al . Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021;371:602–9.
- Davar D , Dzutsev AK , McCulloch JA , Rodrigues RR , Chauvin J-M , Morrison RM et al . Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021;371:595–602.
- Griffin ME , Espinosa J , Becker JL , Luo J-D , Carroll TS , Jha JK et al . Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 2021;373:1040–6.
- Mager LF , Burkhard R , Pett N , Cooke NCA , Brown K , Ramay H et al . Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369:1481–9.
- Ansaldo E , Belkaid Y . How microbiota improve immunotherapy. Science 2021;373:966–7.
- Ansaldo E , Farley TK , Belkaid Y . Control of immunity by the microbiota. Annu Rev Immunol 2021;39:449–79.
- Zhang Q , Ma C , Duan Y , Heinrich B , Rosato U , Diggs LP et al . Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov 2021;11:1248–67.
- Ding T , Schloss PD . Dynamics and associations of microbial community types across the human body. Nature 2014;509:357–60.
- Byrd AL , Liu M , Fujimura KE , Lyalina S , Nagarkar DR , Charbit B et al . Gut microbiome stability and dynamics in healthy donors and patients with non-gastrointestinal cancers. J Exp Med 2021;218:e20200606.
- Healy CM , Moran GP . The microbiome and oral cancer: more questions than answers. Oral Oncol 2019;89:30-3.
- Swaney MH , Kalan LR . Living in your skin: microbes, molecules and mechanisms. Infect Immun 2021;89:e00695–20.
- Willis JR , Gabaldón T . The human oral microbiome in health and disease: from sequences to ecosystems. Microorganisms 2020;8:308.
- Xu J , Peng J-J , Yang W , Fu K , Zhang Y . Vaginal microbiomes and ovarian cancer: a review. Am J Cancer Res 2020;10:743–56.
- Nejman D , Livyatan I , Fuks G , Gavert N , Zwang Y , Geller LT et al . The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80.
- Jin C , Lagoudas GK , Zhao C , Bullman S , Bhutkar A , Hu B et al . Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019;176:998–1013.
- Pushalkar S , Hundeyin M , Daley D , Zambirinis CP , Kurz E , Mishra A et al . The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 2018;8:403–16.
- McAllister F , Khan MAW , Helmink B , Wargo JA . The tumor microbiome in pancreatic cancer: bacteria and beyond. Cancer Cell 2019;36:577–9.
- Kadosh E , Snir-Alkalay I , Venkatachalam A , May S , Lasry A , Elyada E et al . The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020;586:133–8.
- Birch J , Gil J . Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34:1565–76.
- Faget DV , Ren Q , Stewart SA . Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 2019;19:439–53.
- Gorgoulis V , Adams PD , Alimonti A , Bennett DC , Bischof O , Bishop C et al . Cellular senescence: defining a path forward. Cell 2019;179:813–27.
- He S , Sharpless NE . Senescence in health and disease. Cell 2017;169:1000–11.
- Kowald A , Passos JF , Kirkwood TBL . On the evolution of cellular senescence. Aging Cell 2020;19:e13270.
- Lee S , Schmitt CA . The dynamic nature of senescence in cancer. Nat Cell Biol 2019;21:94–101.
- Wang B , Kohli J , Demaria M . Senescent cells in cancer therapy: friends or foes? Trends Cancer 2020;6:838–57.
- Baker DJ , Childs BG , Durik M , Wijers ME , Sieben CJ , Zhong J et al . Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–9.
- Ruhland MK , Loza AJ , Capietto A-H , Luo X , Knolhoff BL , Flanagan KC et al . Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 2016;7:11762.
- Hwang HJ , Lee Y-R , Kang D , Lee HC , Seo HR , Ryu J-K et al . Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett 2020;490:100–10.
- Wang D , Xiao F , Feng Z , Li M , Kong L , Huang L et al . Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res 2020;22:103.
- Amor C , Feucht J , Leibold J , Ho Y-J , Zhu C , Alonso-Curbelo D et al . Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020;583:127–32.
- Aster JC , Pear WS , Blacklow SC . The varied roles of notch in cancer. Annu Rev Pathol 2017;12:245–75.
- Kuczynski EA , Vermeulen PB , Pezzella F , Kerbel RS , Reynolds AR . Vessel co-option in cancer. Nat Rev Clin Oncol 2019;16:469–93.
Google ScholarCrossref
[/toggle]